1.本发明涉及抗病毒药物的制备,具体涉及用于合成帕罗韦德的中间产物及制备方法。
背景技术:
2.据美国食品和药物管理局(fda)当地时间12月22日消息,辉瑞公司的口服新冠药“paxlovid”当天成为了美国首个获批的口服抗新冠病毒药物。报道称,“paxlovid”用于治疗轻中度新冠肺炎,适用人群为12岁以上、体重40公斤以上的高危患者。paxlovid是一种复方制剂,由300mg(2片150mg)nirmatrelvir和一片100mg利托那韦组成,paxlovid将住院或死亡风险降低89%(症状出现后3天内启动治疗)和88%(症状出现后5天内启动治疗)。paxlovid活性药物成分中,nirmatrelvir是一种源于辉瑞实验室的新型主蛋白酶(mpro,也被称为3cl蛋白酶)抑制剂,专门设计用于阻断sars-cov-2mpro的活性,这种酶是冠状病毒复制所需要的。低剂量利托那韦有助于减缓nirmatrelvir的代谢或分解,使其在较高浓度下在体内保持较长时间的活性,以帮助对抗病毒。
3.现有的帕罗韦德(pf-07321332)的合成方法,使用大量缩合剂存在不同程度的消旋风险,甚至引入多种代谢杂质,后处理相对复杂。
技术实现要素:
4.本发明的目的在于解决现有技术的不足,设计出合成路线中关键的中间体化合物,简化合成路线,降低消旋风险,简化反应后处理步骤。
5.为了实现上述目的,设计一种用于合成帕罗韦德(pf-07321332)的化合物,所述化合物的分子结构为
6.一种上述用于合成帕罗韦德的化合物的制备方法,所述化合物制备路线如下:
[0007][0008]
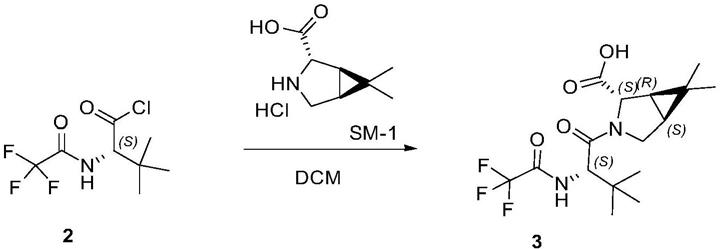
一种用于合成帕罗韦德中间产物的制备方法,所述中间产物由上述的化合物合成,由化合物与sm-1反应后制得第二化合物,第二化合物再通过与二氯亚砜反应制备中间产物,制备路线如下:
[0009]
第一步,
[0010]
第二步,
[0011]
一种由上述方法直接制得的中间产物,所述中间产物分子结构如下:
[0012][0013]
一种应用上述中间产物合成帕罗韦德的方法,合成方法具体如下:
[0014][0015]
与现有技术相比,本发明的优点如下:将羧酸直接做成酰氯,做出关键的中间体,降低消旋风险,简化后处理操作,提高收率。
具体实施方式
[0016]
以下结合实施例对于本发明做进一步说明,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明的保护范围。
[0017]
步骤1
[0018]
反应方程式:
[0019][0020]
原料使用一览表:
[0021][0022][0023]
实验操作:
[0024]
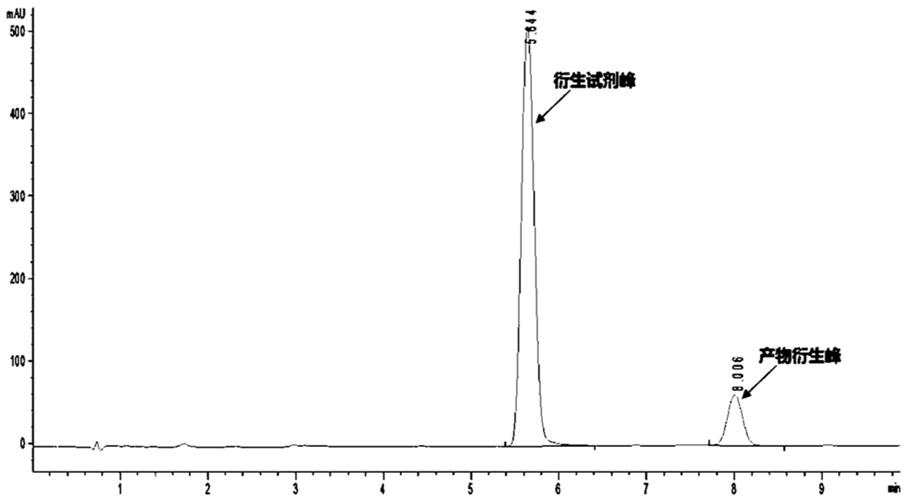
反应釜中,氮气保护下加入化合物1(1.00kg,4.4mol),二氯甲烷(dcm,10.0l)。反应液降温至-5-0℃,缓慢滴加二氯亚砜(socl2,1.00kg,8.8mol),控制反应体系内温度低于10℃。滴加完毕后,将体系温度调至20-25℃,搅拌4-5h。通过高效液相色谱(hplc)监控反应进度。
[0025]
待反应完全后,将反应液减压蒸干至无液体滴出。蒸干得到的黄色油状物化合物1.05kg,直接用于下一步。
[0026]
步骤2
[0027]
反应方程式:
[0028][0029]
原料使用一览表:
[0030]
materialsmw(g/mol)weight(kg)moles(mol)eq.化合物2245.631.054.271.05sm-1191.660.784.071.0dipea129.241.5812.213.0dcm\15.6l\20vol
[0031]
实验操作:
[0032]
将sm-1(0.78kg,4.07mol)溶于二氯甲烷(dcm,15.6l)中,氮气保护下加入n,n-二异丙基乙胺(dipea,1.58kg,12.21mol)。反应液降温至0℃,化合物2(第二化合物,1.05kg,4.27mol)溶于dcm(2.1l)中,缓慢滴入反应液,控制反应体系内温度低于5℃。滴加完毕后于
0℃下搅拌30min,通过高效液相色谱(hplc)监控反应进程。
[0033]
待反应完全后,将反应液倒入1n盐酸溶液(14l)中,搅拌均匀,静置待反应液分层,分离出有机相。有机相用饱和氯化钠溶液(10l)洗涤后,加入无水硫酸钠干燥,分离出有机相,蒸干。
[0034]
用乙酸乙酯,正庚烷搅拌结晶得白色固体化合物3(1.29kg,两步yield:80%)。
[0035]
步骤3
[0036]
反应方程式:
[0037][0038]
原料使用一览表:
[0039]
materialsmw(g/mol)weight(kg)moles(mol)eq.化合物3364.361.193.261.0socl2118.970.786.522.0dcm\10.0l\10vol
[0040]
实验操作:
[0041]
反应釜中,氮气保护下加入化合物3(第三化合物,1.19kg,3.26mol),dcm(10.0l)。反应液降温至-5-0℃,滴加socl2(0.78kg,6.52mol),控制反应体系内温度低于10℃。滴加完毕,将体系温度调至20-25℃,搅拌4-5h,通过高效液相色谱(hplc)监控反应进程。
[0042]
反应完毕后,反应液减压蒸干至无液体滴出。蒸干得黄色油状物化合物1.25kg,直接用于下一步。
[0043]
步骤4
[0044]
反应方程式:
[0045][0046]
原料使用一览表:
[0047]
materialsmw(g/mol)weight(kg)moles(mol)eq.化合物4382.131.253.271.00sm-2207.660.723.431.05
dipea129.241.279.813.00dcm\25l\20vol
[0048]
实验操作:
[0049]
将sm-2(0.72kg,3.43mol)溶于dcm(25l)中,氮气保护下加入dipea(1.27kg,9.81mol)。调节反应体系温度降至0℃,化合物4(1.25kg,3.27mol)溶于dcm(2.5l),缓慢滴入反应液,控制反应体系内温低于5℃。滴加完毕后于0℃下搅拌30min,通过高效液相色谱(hplc)监控反应进程。
[0050]
待反应完全后,将反应液倒入1n盐酸溶液(12l)中,搅拌均匀,静置待反应液分层,分离出有机相。有机相用饱和氯化钠溶液(10l)洗涤后,加入无水硫酸钠干燥,分离出有机相,蒸干。
[0051]
用乙酸乙酯,正庚烷搅拌结晶得白色固体化合物5(1.46kg,两步yield:86%)。
[0052]
步骤5
[0053]
反应方程式
[0054][0055]
原料使用一览表:
[0056]
materialsmw(g/mol)weight(kg)moles(mol)eq.化合物5517.541.452.81.00tfaa210.031.768.43.0et3n101.191.7016.86.00dcm\29l\20vol
[0057]
实验操作:
[0058]
将化合物(51.45kg,2.8mol)溶于dcm(29l)中,氮气保护,加入三乙胺(et3n,1.70kg,16.8mol)。反应液降温至-10-0℃,三氟醋酸酐(tfaa,1.76kg,8.4mol)缓慢滴入反应液中,控制反应体系内温低于5℃。滴加完毕后于0℃下搅拌30min,通过高效液相色谱(hplc)监控反应进程。
[0059]
待反应完全后,将反应液倒入1n氯化铵溶液(12l)中,搅拌均匀,静置待反应液分层,分离出有机相。有机相用饱和氯化钠溶液(10l)洗涤后,加入无水硫酸钠干燥,分离出有机相,蒸干。
[0060]
用乙酸乙酯,正庚烷搅拌结晶得白色固体化合物6(1.18kg,yield:84%)。
[0061]
通过设计出关键的中间产物化合物2,化合物4,可以简化整个合成步骤;通过二氯亚砜将羧基变成酰氯,可以降低消旋风险,简化后处理操作,提高产率。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。