1.本发明涉及化学药物合成技术领域,尤其涉及一种芳基吡唑类化合物的制备方法。
背景技术:
2.吡唑类化合物是一种非常重要的含氮五元杂环化合物,它是很多生物活性分子和药物(如非泼罗尼、塞来西布、利莫那班等)的重要骨架。尤其是含氟吡唑类化合物具有广谱的杀虫活性,活性高,应用范围广,如非泼罗尼,可以通过阻断γ-氨基丁酸所受体控制的神经膜的氯离子通道诱导氯离子流,引起神经系统极度兴奋而导致虫体死亡,对多种蚤类、蜱类等节肢类动物具有良好的防控效果。而且,有文献报道(wo9824767,ep933363,ep957094,cn1893825a)至少有一个氟连接至环丙基环的1-芳基-4-环丙基吡唑相对于现有的抗寄生虫化合物、药物具有更好的活性和长效作用。但是,含有氟取代的环丙基化合物在工业化生产合成上仍然存在一定的限制,亟需对其合成方法进行优化改良。
3.抗寄生虫化合物5-氨基-3-氰基-1-[芳基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑的结构通式如化合物i所示:
[0004][0005]
已报道的文献中,以化合物5-氨基-3-氰基-1-[芳基]-吡唑(1)为起始原料合成5-氨基-3-氰基-1-[芳基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑(化合物i)的主要有以下两条合成路线(us 20060014802a1):
[0006]
合成路线一通式:
[0007][0008]
合成路线二通式:
[0009][0010]
上述合成路线方案中,均需要使用到昂贵且危险的化合物重氮甲烷,这并不利于工业化大生产;而且,上述路线中,后处理大都需要通过柱层析等较为麻烦的纯化手段,这在大规模生产条件下具有很大的局限性。
[0011]
因此,亟需研究发展一种反应高效,操作简便,物料安全易得,后处理简洁,适合大规模生产的新合成工艺。
技术实现要素:
[0012]
本发明的目的在于克服现有技术的缺陷,提供一种芳基吡唑类化合物的制备方法。
[0013]
为了实现上述发明目的,本发明提供以下技术方案:
[0014]
本发明提供了一种芳基吡唑类化合物的制备方法,包含下列步骤:
[0015]
(1)将5-氨基-3-氰基-1-芳基吡唑、三氟乙酸酐、酸和溶剂混合进行水解反应,得到中间体2;
[0016]
(2)将中间体2、n,n-二甲基甲酰胺二甲基缩醛和溶剂混合进行缩合反应,得到中间体3;
[0017]
(3)在保护气氛中,将中间体3、甲基三苯基磷盐、碱和溶剂混合进行叶立德反应,得到中间体4;
[0018]
(4)在保护气氛中,将中间体4、二氟卡宾试剂、催化剂和溶剂混合进行环丙烷化反应,得到中间体5;
[0019]
(5)将中间体5、酸和溶剂混合进行水解反应,即得所述芳基吡唑类化合物;
[0020]
所述步骤(1)中5-氨基-3-氰基-1-芳基吡唑的结构如下:
[0021][0022]
所述r为卤素、烷基、苯基、含氟烷基或氰基,所述r为单取代、双取代、三取代、四取代或五取代。
[0023]
作为优选,步骤(1)中所述酸为盐酸、甲酸、乙酸、硫酸、对甲苯磺酸或苯甲酸;
[0024]
所述溶剂为n,n-二甲基甲酰胺、二甲基亚砜、甲苯、四氢呋喃、二氯甲烷、乙腈或1,4-二氧六环;
[0025]
所述5-氨基-3-氰基-1-芳基吡唑和三氟乙酸酐的摩尔比为1:2~3.5;
[0026]
所述5-氨基-3-氰基-1-芳基吡唑和酸的摩尔比为1:2~3.5;
[0027]
所述5-氨基-3-氰基-1-芳基吡唑和溶剂的摩尔体积比为1mol:1~1.2l。
[0028]
作为优选,步骤(1)中所述水解反应的温度为20~110℃;所述水解反应的时间为1~1.5h。
[0029]
作为优选,所述步骤(2)中溶剂为n,n-二甲基甲酰胺、甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或二甲苯;
[0030]
所述中间体2和n,n-二甲基甲酰胺二甲基缩醛的摩尔比为1:1.2~2.0;
[0031]
所述中间体2和溶剂的摩尔体积比为1mol:2.4~2.6l。
[0032]
作为优选,所述步骤(2)中缩合反应的温度为20~110℃,时间为3~4h。
[0033]
作为优选,所述步骤(3)中甲基三苯基磷盐为甲基三苯基溴化磷、甲基三苯基氯化磷或甲基三苯基碘化磷;
[0034]
所述碱为碳酸钾、丁基锂、叔丁醇钾、氢氧化钠、叔丁醇锂或叔丁醇钠;
[0035]
所述溶剂为甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或n,n-二甲基甲酰胺;
[0036]
所述中间体3和甲基三苯基磷盐的摩尔比为1:1.5~2.0;
[0037]
所述中间体3和碱的摩尔比为1:1.5~2.0;
[0038]
所述中间体3和溶剂的摩尔体积比为1mol:9.2~9.8l。
[0039]
作为优选,所述步骤(3)中叶立德反应的温度为20~30℃,时间为2~3h。
[0040]
作为优选,所述步骤(4)中二氟卡宾试剂为氟磺酰基二氟乙酸甲酯、二氟氯乙酸钠、三氟甲基三甲基硅烷或三甲基硅烷基2-(氟磺酰基)二氟乙酸酯;
[0041]
所述催化剂为碘化钾、碘化钠、四正丁基铵二氟代三苯基硅酸盐、碘化锂、氟化钠或氟化钾;
[0042]
所述溶剂为甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或n,n-二甲基甲酰胺;
[0043]
所述中间体4和二氟卡宾试剂的摩尔比为1:3~6;
[0044]
所述中间体4和催化剂的摩尔比为1:0.3~0.6;
[0045]
所述中间体4和溶剂的摩尔体积比为1mol:0.5~0.8l。
[0046]
作为优选,所述步骤(4)中环丙烷化反应的温度为80~130℃,所述环丙烷化反应的时间为24~28h。
[0047]
作为优选,步骤(5)中所述酸为盐酸、甲酸、乙酸、硫酸、对甲苯磺酸或苯甲酸;
[0048]
所述溶剂为甲醇、乙醇、异丙醇、乙腈、二氯甲烷或四氢呋喃;
[0049]
所述中间体5和酸的摩尔比为1:1.5~5.0;
[0050]
所述中间体5和溶剂的摩尔体积比为1mol:6~7l;
[0051]
所述步骤(5)中水解反应的温度为20~90℃,时间为12~14h。
[0052]
本发明具有以下优点:
[0053]
(1)本发明提供的制备方法简单,相对于已有工艺路线中需要柱层析等复杂的纯化手段,本发明中所有中间体以及最终产物均可以通过简单的重结晶获得高纯度的目标化合物,极大的降低了后处理的难度,提高了工艺的应用性。尤其是合成中间体2和中间体3,反应完成后通过简单的重结晶操作即可获得高收率(》95%),高纯度(》99%)的目标化合物。
[0054]
(2)本发明使用的物料安全,便宜易得,能够有效降低生产成本且提高大规模生产安全性,改善了反应过程中化合物的纯化难题,为此类抗寄生虫活性化合物的大规模生产、应用奠定了基础。
附图说明
[0055]
图1为实施例1制备得到中间体2的核磁共振氢谱图;
[0056]
图2为实施例1制备得到中间体3的核磁共振氢谱图;
[0057]
图3为实施例1制备得到中间体4的核磁共振氢谱图;
[0058]
图4为实施例1制备得到中间体5的核磁共振氢谱图;
[0059]
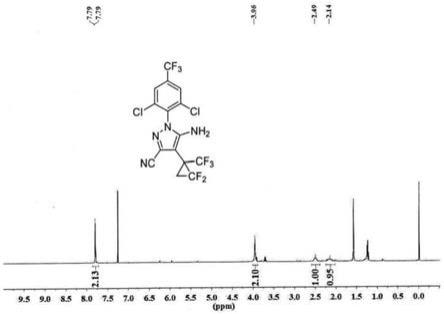
图5为实施例1制备得到5-氨基-3-氰基-1-[2,6-二氯-4-(三氟甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑的核磁共振氢谱图。
具体实施方式
[0060]
本发明提供了一种芳基吡唑类化合物的制备方法,包含下列步骤:
[0061]
(1)将5-氨基-3-氰基-1-芳基吡唑、三氟乙酸酐、酸和溶剂混合进行水解反应,得到中间体2;
[0062]
(2)将中间体2、n,n-二甲基甲酰胺二甲基缩醛和溶剂混合进行缩合反应,得到中间体3;
[0063]
(3)在保护气氛中,将中间体3、甲基三苯基磷盐、碱和溶剂混合进行叶立德反应,得到中间体4;
[0064]
(4)在保护气氛中,将中间体4、二氟卡宾试剂、催化剂和溶剂混合进行环丙烷化反应,得到中间体5;
[0065]
(5)将中间体5、酸和溶剂混合进行水解反应,即得所述芳基吡唑类化合物;
[0066]
所述步骤(1)中5-氨基-3-氰基-1-芳基吡唑的结构如下:
[0067][0068]
所述r为卤素、烷基、苯基、含氟烷基或氰基,所述r为单取代、双取代、三取代、四取代或五取代。
[0069]
在本发明中,步骤(1)中所述酸优选为盐酸、甲酸、乙酸、硫酸、对甲苯磺酸或苯甲酸。
[0070]
在本发明中,当选择盐酸或硫酸时,将盐酸或硫酸配制成水溶液,盐酸水溶液的质量分数优选为10~12%,进一步优选为10.4~11.6%,更优选为10.8~11.2%;硫酸水溶液的质量分数优选为10~12%,进一步优选为10.4~11.6%,更优选为10.8~11.2%。
[0071]
在本发明中,所述溶剂优选为n,n-二甲基甲酰胺、二甲基亚砜、甲苯、四氢呋喃、二氯甲烷、乙腈或1,4-二氧六环。
[0072]
在本发明中,所述5-氨基-3-氰基-1-芳基吡唑和三氟乙酸酐的摩尔比优选为1:2~3.5,进一步优选为1:2.5~3,更优选为1:2.6~2.9。
[0073]
在本发明中,所述5-氨基-3-氰基-1-芳基吡唑和酸的摩尔比优选为1:2~3.5,进一步优选为1:2.5~3,更优选为1:2.6~2.9。
[0074]
在本发明中,所述5-氨基-3-氰基-1-芳基吡唑和溶剂的摩尔体积比优选为1mol:1~1.2l,进一步优选为1mol:1.05~1.15l,更优选为1mol:1.08~1.12l。
[0075]
在本发明中,将5-氨基-3-氰基-1-芳基吡唑和溶剂在搅拌状态下混合,然后滴加三氟乙酸酐,滴加完毕后进行酰化反应。
[0076]
在本发明中,所述搅拌无具体要求,混合均匀即可;所述滴加的速率优选为15~25min/mol,进一步优选为16~24min/mol,更优选为18~22min/mol。
[0077]
在本发明中,所述酰化反应的温度优选为20~80℃,进一步优选为30~70℃,更优选为40~60℃;所述酰化反应的时间优选为2.5~3.5h,进一步优选为2.6~3.4h,更优选为2.8~3.2h。
[0078]
在本发明中,酰化反应结束后将酸加入到反应体系中进行下一步的水解反应。
[0079]
在本发明中,步骤(1)中所述水解反应的温度优选为20~110℃,进一步优选为40~90℃,更优选为60~70℃;所述水解反应的时间优选为1~1.5h,进一步优选为1.1~1.4h,更优选为1.2~1.3h。
[0080]
在本发明中,采用hplc或tlc检测水解反应是否完成,所述hplc检测优选使用c18柱,乙腈和水作为流动相;tlc检测使用的展开剂优选为石油醚和乙酸乙酯,乙酸乙酯和石油醚的体积比优选为1:2~5,进一步优选为1:3~4,更优选为1:3.4~3.6。
[0081]
在本发明中,所述水解反应完成后进行后处理,所述后处理优选为顺次进行的减压蒸馏去除溶剂、重结晶和抽滤、洗涤和烘干,即得所述中间体2。
[0082]
在本发明中,所述步骤(1)的反应过程如下:
[0083][0084]
在本发明中,所述步骤(2)中溶剂优选为n,n-二甲基甲酰胺、甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或二甲苯。
[0085]
在本发明中,所述中间体2和n,n-二甲基甲酰胺二甲基缩醛的摩尔比优选为1:1.2~2.0,进一步优选为1:1.4~1.8,更优选为1:1.5~1.7。
[0086]
在本发明中,所述中间体2和溶剂的摩尔体积比优选为1mol:2.4~2.6l,进一步优选为1mol:2.44~2.56l,更优选为1mol:2.48~2.52l。
[0087]
在本发明中,将中间体2和溶剂混合后,向混合体系中滴加n,n-二甲基甲酰胺二甲基缩醛,所述滴加的速率优选为30~40min/mol,进一步优选为32~38min/mol,更优选为34~36min/mol;滴加完毕后进行缩合反应。
[0088]
在本发明中,所述步骤(2)中缩合反应的温度优选为20~110℃,进一步优选为40~90℃,更优选为60~70℃;时间优选为3~4h,进一步优选为3.2~3.8h,更优选为3.4~3.6h。
[0089]
在本发明中,采用hplc或tlc检测缩合反应是否完成,所述hplc检测优选使用c18柱,乙腈和水作为流动相;tlc检测使用的展开剂优选为石油醚和乙酸乙酯,乙酸乙酯和石油醚的体积比优选为1:2~5,进一步优选为1:3~4,更优选为1:3.4~3.6。
[0090]
在本发明中,所述缩合反应完成后进行后处理,所述后处理优选为顺次进行的减压蒸馏去除溶剂、重结晶和抽滤、洗涤和烘干,即得所述中间体3;所述重结晶的体系优选为乙酸乙酯-石油醚,所述洗涤的试剂优选为石油醚。
[0091]
在本发明中,所述步骤(2)的反应过程如下:
[0092][0093]
在本发明中,所述步骤(3)中甲基三苯基磷盐优选为甲基三苯基溴化磷、甲基三苯基氯化磷或甲基三苯基碘化磷。
[0094]
在本发明中,所述碱优选为碳酸钾、丁基锂、叔丁醇钾、氢氧化钠、叔丁醇锂或叔丁醇钠。
[0095]
在本发明中,所述溶剂优选为甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或n,n-二甲基甲酰胺。
[0096]
在本发明中,所述中间体3和甲基三苯基磷盐的摩尔比优选为1:1.5~2.0,进一步优选为1:1.6~1.9,更优选为1:1.7~1.8。
[0097]
在本发明中,所述中间体3和碱的摩尔比优选为1:1.5~2.0,进一步优选为1:1.6
~1.9,更优选为1:1.7~1.8。
[0098]
在本发明中,所述中间体3和溶剂的摩尔体积比优选为1mol:9.2~9.8l,进一步优选为1mol:9.3~9.7l,更优选为1mol:9.4~9.6l。
[0099]
在本发明中,所述步骤(3)中保护气氛优选为氮气、氦气或氩气。
[0100]
在本发明中,将甲基三苯基锂盐和第一部分溶剂混合,第一部分溶剂的体积优选为全部溶剂体积的75~80%,进一步优选为76~79%,更优选为77~78%;所述混合的方式优选为搅拌,搅拌的转速无明确要求,能混合均匀即可;所述搅拌的时间优选为4~6h,进一步优选为4.5~5.5h,更优选为4.8~5.2h;所述搅拌的温度优选为-2~2℃,进一步优选为-1~1℃,更优选为0℃;搅拌结束后得到混合体系;将碱加入到混合体系中进行活化,所述活化的温度优选为20~30℃,进一步优选为22~28℃,更优选为24~26℃,所述活化的时间优选为5~7h,进一步优选为5.4~6.6h,更优选为5.8~6.2h;活化结束后降温,所述降温的目标温度优选为-2~2℃,进一步优选为-1~1℃,更优选为0℃,降至目标温度后得到反应体系,将中间体3和剩余溶剂混合后加入到反应体系中,进行叶立德反应。
[0101]
在本发明中,所述步骤(3)中叶立德反应的温度优选为20~30℃,进一步优选为22~28℃,更优选为24~26℃;时间优选为2~3h,进一步优选为2.2~2.8h,更优选为2.4~2.6h。
[0102]
在本发明中,采用hplc或tlc检测叶立德反应是否完成,所述hplc检测优选使用c18柱,乙腈和水作为流动相;tlc检测使用的展开剂优选为石油醚和乙酸乙酯,乙酸乙酯和石油醚的体积比优选为1:2~5,进一步优选为1:3~4,更优选为1:3.4~3.6。
[0103]
在本发明中,所述叶立德反应完成后,顺次进行淬灭、减压蒸馏、萃取、重结晶、抽滤、洗涤和干燥,即得所述中间体4。所述淬灭的试剂优选为水、饱和氯化铵水溶液、饱和碳酸钠水溶液或饱和碳酸氢钠水溶液;所述萃取的溶剂优选为乙酸乙酯、二氯甲烷、乙醚或甲基叔丁基醚;所述重结晶的溶剂优选为乙酸乙酯、二氯甲烷、甲醇、乙醇、异丙醇、石油醚、正己烷、环己烷、甲基叔丁基醚、丙酮、n,n-二甲基甲酰胺、二甲基亚砜、甲苯和水中的一种或几种。
[0104]
在本发明中,所述步骤(3)的反应过程如下:
[0105][0106]
在本发明中,所述步骤(4)中二氟卡宾试剂优选为氟磺酰基二氟乙酸甲酯、二氟氯乙酸钠、三氟甲基三甲基硅烷或三甲基硅烷基2-(氟磺酰基)二氟乙酸酯。
[0107]
在本发明中,所述催化剂优选为碘化钾、碘化钠、四正丁基铵二氟代三苯基硅酸盐、碘化锂、氟化钠或氟化钾。
[0108]
在本发明中,所述溶剂优选为甲苯、四氢呋喃、二氯甲烷、乙腈、1,4-二氧六环或n,n-二甲基甲酰胺。
[0109]
在本发明中,所述中间体4和二氟卡宾试剂的摩尔比优选为1:3~6,进一步优选为1:4~5,更优选为1:4.4~4.6。
[0110]
在本发明中,所述中间体4和催化剂的摩尔比优选为1:0.3~0.6,进一步优选为1:0.4~0.5,更优选为1:0.44~0.46。
[0111]
在本发明中,所述中间体4和溶剂的摩尔体积比优选为1mol:0.5~0.8l,进一步优选为1mol:0.6~0.7l,更优选为1mol:0.64~0.66l。
[0112]
在本发明中,所述步骤(4)中保护气氛优选为氮气、氦气或氩气。
[0113]
在本发明中,将中间体、催化剂、溶剂和二氟卡宾试剂混合进行环丙烷化反应。
[0114]
在本发明中,所述步骤(4)中环丙烷化反应的温度优选为80~130℃,进一步优选为90~120℃,更优选为100~110℃;所述环丙烷化反应的时间优选为24~28h,进一步优选为25~27h,更优选为25.5~26.5h。
[0115]
在本发明中,采用hplc或tlc检测环丙烷化反应是否完成,所述hplc检测优选使用c18柱,乙腈和水作为流动相;tlc检测使用的展开剂优选为石油醚和乙酸乙酯,乙酸乙酯和石油醚的体积比优选为1:2~5,进一步优选为1:3~4,更优选为1:3.4~3.6。
[0116]
在本发明中,所述环丙烷化反应完成后,顺次进行淬灭、减压蒸馏、萃取、重结晶、洗涤和干燥,即得所述中间体5;所述淬灭的试剂优选为水、饱和氯化铵水溶液、饱和碳酸钠水溶液或饱和碳酸氢钠水溶液;所述萃取的溶剂优选为乙酸乙酯、二氯甲烷、乙醚或甲基叔丁基醚;所述重结晶的溶剂优选为乙酸乙酯、二氯甲烷、甲醇、乙醇、异丙醇、石油醚、正己烷、环己烷、甲基叔丁基醚、丙酮、n,n-二甲基甲酰胺、二甲基亚砜、甲苯和水中的一种或几种。
[0117]
在本发明中,所述步骤(4)的反应过程如下:
[0118][0119]
在本发明中,步骤(5)中所述酸优选为盐酸、甲酸、乙酸、硫酸、对甲苯磺酸或苯甲酸。
[0120]
在本发明中,所述溶剂优选为甲醇、乙醇、异丙醇、乙腈、二氯甲烷或四氢呋喃。
[0121]
在本发明中,所述中间体5和酸的摩尔比优选为1:1.5~5.0,进一步优选为1:2~4.5,更优选为1:2.5~4。
[0122]
在本发明中,所述中间体5和溶剂的摩尔体积比优选为1mol:6~7l,进一步优选为1mol:6.2~6.8l,更优选为1mol:6.4~6.6l。
[0123]
在本发明中,将中间体5、酸和溶剂混合进行水解反应。
[0124]
在本发明中,所述步骤(5)中水解反应的温度优选为20~90℃,进一步优选为30~80℃,更优选为50~60℃;时间优选为12~14h,进一步优选为12.5~13.5h,更优选为12.9~13.1h。
[0125]
在本发明中,采用hplc或tlc检测水解反应是否完成,所述hplc检测优选使用c18柱,乙腈和水作为流动相;tlc检测使用的展开剂优选为石油醚和乙酸乙酯,乙酸乙酯和石油醚的体积比优选为1:2~5,进一步优选为1:3~4,更优选为1:3.4~3.6。
[0126]
在本发明中,步骤(5)中水解反应完成后,顺次进行淬灭、减压蒸馏、萃取、洗涤和干燥,即得所述芳基吡唑类化合物。所述淬灭的试剂优选为水、饱和氯化铵水溶液、饱和碳酸钠水溶液或饱和碳酸氢钠水溶液;所述萃取的溶剂优选为乙酸乙酯、二氯甲烷、乙醚或甲基叔丁基醚;所述重结晶的溶剂优选为乙酸乙酯、二氯甲烷、甲醇、乙醇、异丙醇、石油醚、正己烷、环己烷、甲基叔丁基醚、丙酮、n,n-二甲基甲酰胺、二甲基亚砜、甲苯和水中的一种或几种。
[0127]
在本发明中,所述步骤(5)的反应过程如下:
[0128][0129]
在本发明中,制备得到的芳基吡唑类化合物优选为:5-氨基-3-氰基-1-(苯基)-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑、5-氨基-3-氰基-1-[2,6-二氯-4-(三氟甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑、5-氨基-3-氰基-1-[2,6-二氯苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑、5-氨基-3-氰基-1-[4-(甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑、5-氨基-3-氰基-1-[4-(氰基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑。
[0130]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0131]
实施例1
[0132]
将0.5mol5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基苯基)吡唑溶于500.0ml的四氢呋喃中,在搅拌状态下将1.5mol的三氟乙酸酐用滴液漏斗于30min滴加进搅拌着的反应体系中。滴加完成后,在50℃下反应3h完成酰化反应。随后,将523.0ml、10%稀盐酸加入反应体系中,在105℃下水解反应1h。采用c18柱、乙腈和水作为流动相进行hplc检测,水解反应完成后,减压蒸馏除去溶剂四氢呋喃,降温重结晶,析出的固体抽滤,大量水洗涤,固体置于鼓风烘箱中干燥,得到白色固体即为中间体2,质量为198.0g,产率95%,纯度99.3%。将中间体2进行核磁共振,得到的氢谱如图1所示,1h nmr(400mhz,cdcl3)δ7.84(s,2h),6.13(brs,2h).hrms(esi):calcd for c
13
h5cl2f6n4o(m h
)416.9739;found 416.9743。
[0133]
将0.5mol中间体2溶于1200.0ml二氯甲烷中,然后将0.75mol的n,n-二甲基甲酰胺二甲基缩醛用滴液漏斗于25min滴加进搅拌着的反应体系中。滴加完成后,在40℃下缩合反应3h。采用c18柱、乙腈和水作为流动相进行hplc检测,缩合反应完成后,减压蒸馏除去溶剂,残余物用乙酸乙酯-石油醚体系重结晶,然后抽滤,石油醚洗涤,固体置于鼓风烘箱中干燥,得到白色固体,即为中间体3,质量为226.0g,产率96%,纯度99%。将中间体3进行核磁共振,得到的氢谱如图2所示,1h nmr(400mhz,cdcl3)δ8.38(s,1h),7.73(s,2h),3.15(s,3h),2.85(s,3h).hrms(esi):calcd for c
13
h5cl2f6n4o(m h
)472.0161;found 472.0156。
[0134]
将0.1mol甲基三苯基碘化磷加入1000ml烧瓶中,氮气置换,使瓶中为氮气氛围,然后加入360.0ml1,4-二氧六环,在0℃下均匀搅拌搅拌5h。将0.1mol叔丁醇锂加入混合体系。
然后,在25℃下活化反应6h。将烧瓶降温至0℃,随后将0.05mol中间体3溶于100.0ml1,4-二氧六环中,缓慢加入混合体系,在25℃中反应2h。采用c18柱、乙腈和水作为流动相进行hplc检测,叶立德反应完成后,加入饱和氯化铵水溶液淬灭反应,减压蒸馏除去1,4-二氧六环,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,洗涤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体4,质量为15.2g,产率65%,纯度98%。将中间体4进行核磁共振,得到的氢谱如图3所示,1h nmr(400mhz,cdcl3)δ7.70(s,2h),7.67(s,1h),6.24(m,1h),5.90(m,1h),2.97(s,3h),2.77(s,3h).hrms(esi):calcd for c
13
h5cl2f6n4o(m h
)470.0368;found 470.0361。
[0135]
将0.2mol中间体4、0.1mol氟化钠加入500ml反应器中,氮气置换,使反应器中为氮气氛围,然后加入100.0ml 1,4-二氧六环,再将1mol三甲基硅烷基2-(氟磺酰基)二氟乙酸酯加入反应器中,在130℃下反应24h。采用c18柱、乙腈和水作为流动相进行hplc检测,反应完成后,加入饱和碳酸氢钠水溶液淬灭反应,减压蒸馏除去1,4-二氧六环,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体5,质量为65.4g,产率63%,纯度97.8%。将中间体5进行核磁共振,得到的氢谱如图4所示。1h nmr(400mhz,dmso-d6)δ8.25(s,1h),8.24(s,1h),7.77(s,1h),3.03-2.92(m,4h),2.72(s,3h),2.08(s,1h).hrms(esi):calcd for c
13
h5cl2f6n4o(m h
)520.0337;found 520.0331。
[0136]
在2000ml烧瓶中,将0.2mol中间体5、0.6mol苯甲酸溶于1200.0ml甲醇,在60℃下反应12h。采用c18柱、乙腈和水作为流动相进行hplc检测,反应完成后,加入饱和碳酸钠水溶液淬灭反应,然后减压蒸馏,水相用乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去乙酸乙酯,然后用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于真空烘箱中干燥,得到淡黄色固体,即为最终产物5-氨基-3-氰基-1-[2,6-二氯-4-(三氟甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑,质量为78.9g,产率85%,纯度99%。将5-氨基-3-氰基-1-[2,6-二氯-4-(三氟甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑进行核磁共振,得到的氢谱如图5所示。1h nmr(400mhz,cdcl3)δ7.79(s,2h),3.96(brs,2h),2.49(s,1h),2.14(s,1h).hrms(esi):calcd for c
13
h5cl2f6n4o(m h
)464.9915;found 464.9911。
[0137]
实施例2
[0138]
将1mol5-氨基-3-氰基-1-(4-甲基苯基)吡唑溶于1.2l的二氯甲烷中,在搅拌状态下将3.5mol的三氟乙酸酐用滴液漏斗于70min滴加进搅拌着的反应体系中。滴加完成后,在30℃下反应3.5h完成酰化反应。随后,将2mol苯甲酸加入反应体系中,在60℃下水解反应1.2h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:3.5,水解反应完成后,减压蒸馏除去溶剂二氯甲烷,降温重结晶,析出的固体抽滤,大量水洗涤,固体置于鼓风烘箱中干燥,得到白色固体即为中间体2,产率95.1%,纯度99.4%。
[0139]
将1mol中间体2溶于2.5ln,n-二甲基甲酰胺中,然后将1.8mol的n,n-二甲基甲酰胺二甲基缩醛用滴液漏斗于63min滴加进搅拌着的反应体系中。滴加完成后,在80℃下缩合反应4h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:3.5,缩合反应完成后,减压蒸馏除去溶剂,残余物用乙酸乙酯-石油醚体系重结晶,然后抽滤,石油醚洗涤,固体置于鼓风烘箱中干燥,得到白色固体,即为中间体3,产率95.8%,纯度99.1%。
[0140]
在氮气氛围中,将2mol甲基三苯基溴化磷和7.68l甲苯混合,在0℃下均匀搅拌搅拌5h,将1.8mol碳酸钾加入混合体系中,在30℃下活化5h后降温至0℃,随后将1mol中间体3和1.92l甲苯混合后加入体系中,在30℃反应2h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:3.5,叶立德反应完成后,加入饱和氯化铵水溶液淬灭反应,减压蒸馏除去溶剂,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,洗涤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体4,产率67%,纯度98.5%。
[0141]
在氮气氛围中,将1mol中间体4、0.4mol四正丁基铵二氟代三苯基硅酸盐、0.6l四氢呋喃、4.5mol氟磺酰基二氟乙酸甲酯混合,在105℃下反应26h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:3.5,反应完成后,加入水淬灭反应,减压蒸馏除去溶剂,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体5,产率62.8%,纯度97.6%。
[0142]
将1mol中间体5、3.0mol对甲苯磺酸溶于6.5l乙腈中,在30℃下反应14h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:3.5,反应完成后,加入饱和氯化铵水溶液淬灭反应,然后减压蒸馏,水相用乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏,然后用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于真空烘箱中干燥,得到淡黄色固体,即为最终产物5-氨基-3-氰基-1-[4-(甲基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑,产率86.3%,纯度99.1%。
[0143]
实施例3
[0144]
将0.1mol5-氨基-3-氰基-1-(4-氰基苯基)吡唑溶于0.1l的二甲基亚砜中,在搅拌状态下将0.25mol的三氟乙酸酐用滴液漏斗于5min滴加进搅拌着的反应体系中。滴加完成后,在70℃下反应2.8h完成酰化反应。随后,将0.3mol乙酸加入反应体系中,在100℃下水解反应1.4h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:2,水解反应完成后,减压蒸馏除去溶剂,降温重结晶,析出的固体抽滤,大量水洗涤,固体置于鼓风烘箱中干燥,得到白色固体即为中间体2,产率95.2%,纯度99.2%。
[0145]
将0.1mol中间体2溶于0.24l二甲苯中,然后将0.16mol的n,n-二甲基甲酰胺二甲基缩醛用滴液漏斗于6.4min滴加进搅拌着的反应体系中。滴加完成后,在40℃下缩合反应3h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:4,缩合反应完成后,减压蒸馏除去溶剂,残余物用乙酸乙酯-石油醚体系重结晶,然后抽滤,石油醚洗涤,固体置于鼓风烘箱中干燥,得到白色固体,即为中间体3,产率95.4%,纯度99.3%。
[0146]
在氮气氛围中,将0.175mol甲基三苯基氯化磷和0.705l二氯甲烷混合,在0℃下均匀搅拌4h,将0.18mol丁基锂加入混合体系中,在20℃下活化7h后降温至0℃,随后将0.1mol中间体3和0.235l二氯甲烷混合后加入体系中,在20℃反应3h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:5,叶立德反应完成后,加入饱和碳酸氢钠水溶液淬灭反应,减压蒸馏除去溶剂,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,洗涤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体4,产率69%,纯度99.1%。
[0147]
在氮气氛围中,将0.1mol中间体4、0.03mol碘化锂、0.07l二氯甲烷、0.6mol三氟甲
基三甲基硅烷混合,在95℃下反应27h。采用tlc检测,乙酸乙酯和石油醚的体积比为1:2,反应完成后,加入水淬灭反应,减压蒸馏除去溶剂,乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏除去溶剂,浓缩物使用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于鼓风烘箱中干燥,得到淡黄色固体即为中间体5,产率63.1%,纯度98.2%。
[0148]
将0.1mol中间体5、0.4mol甲酸溶于0.62l异丙醇中,在50℃下反应13h。采用c18柱、乙腈和水作为流动相进行hplc检测,反应完成后,加入饱和碳酸钠水溶液淬灭反应,然后减压蒸馏,水相用乙酸乙酯萃取,有机相用水洗、饱和食盐水洗,干燥。减压蒸馏,然后用乙酸乙酯-石油醚体系重结晶,抽滤,滤饼置于真空烘箱中干燥,得到淡黄色固体,即为最终产物5-氨基-3-氰基-1-[4-(氰基)苯基]-4-[2,2-二氟-1-(三氟甲基)环丙烷]-1h-吡唑,产率86.1%,纯度99.2%。
[0149]
由以上实施例可知,本发明提供了一种芳基吡唑类化合物的制备方法,本发明提供的制备方法简单,相对于已有工艺路线中需要柱层析等复杂的纯化手段,本发明中所有中间体以及最终产物均可以通过简单的重结晶获得高纯度的目标化合物,极大的降低了后处理的难度,提高了工艺的应用性。本发明提供的制备方法得到的芳基吡唑类化合物,产率达到85%,纯度可达99%以上。
[0150]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
