一种生产phb的运动发酵单胞菌的构建方法及其应用
技术领域
1.本发明属于生物技术领域,尤其涉及一种生产phb的运动发酵单胞菌的构建方法及其应用。
背景技术:
2.石油工业生产的塑料具有良好的柔韧性和轻盈等特性,已成为人们生活中不可缺少的商品。然而,石油来源的塑料生物降解性低、回收和分类复杂,存在严重的环境污染,尤其是对海洋生态系统。聚-β-羟丁酸(poly-β-hydroxybutyric,phb)是一类由微生物合成的高分子聚酯,具有生物可降解性和生物相容性,因而被认为是环境友好型材料,广泛应用于农业、环保、生物化工、医药、医用材料等领域。
3.phb的生物合成主要是通过微生物自身代谢,其中最主要的是微生物发酵法,也是研究最为广泛的方法。但是现阶段,大多数的微生物发酵过程是需氧的,需要耗费大量的能量供氧。运动发酵单胞菌(zymomonas mobilis)作为一种兼性厌氧革兰氏阴性菌,具有许多独特的生理特点和优异的工业生产特性,是目前已知唯一能够在厌氧条件下利用2-酮-3-脱氧-6-磷酸葡萄糖酸(entner-doudoroff,ed)途径的微生物,具有较高的糖吸收率、乙醇收率和乙醇耐受性等优良特性。目前已经在运动发酵单胞菌中实现了乳酸、2-3丁二醇,异丁醇等产品的生产。作为一种兼性厌氧生物,运动发酵单胞菌可以在厌氧条件下发酵,且相较于有氧条件表现更好,从而可以解决微生物发酵过程面临的供氧难题,如搅拌、通气以及空气灭菌等。此外,2006年的研究发现在运动发酵单胞菌异源表达phb合成基因,能够积累0.7%的phb,并能在一定程度上加快和促进乙醇的生产。
4.因此,通过基因工程,代谢工程等手段得到运动发酵单胞菌的phb生产菌株,并结合过程工程手段如絮凝和发酵条件优化等方法进一步提高phb产量,使运动发酵单胞菌成为乙醇和phb生产的高效细胞工厂具有重要意义。
技术实现要素:
5.针对现有技术存在的问题,本发明提供了一种生产phb的运动发酵单胞菌的构建方法及其应用,目的在于解决现有技术中的一部分问题或至少缓解现有技术中的一部分问题。
6.本发明是这样实现的,一种生产phb的运动发酵单胞菌的构建方法,包括以下步骤:将序列分别如seq id no.1-seq id no.3所示的phac、phaa和phab,3个外源基因序列按照ptet-phac-rbs-phaa-rbs-phab的顺序串联得到ptet-phacab操纵子,序列如seq id no.6所示;利用基因编辑手段,将zm4基因组上的zmo0038基因用ptet-phacab替换,得到菌株zmpt。
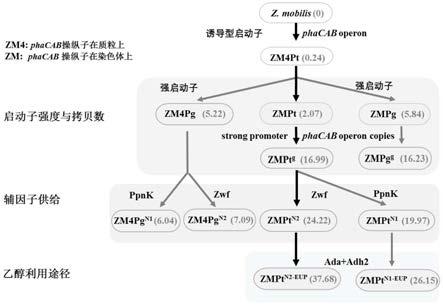
7.与实施例1中构建的zm4pt相比,phacab操纵子位于染色体上比位于质粒中具有更好的生产效果。zm4pt的phb的产量为0.24%dcw,而zmpt的phb的产量为2.07%dcw,产量提高了8.6倍。
8.进一步地,用强启动子pgap替换ptet-phacab中的ptet启动子构建质粒pez-pg,将质粒pez-pg转入zmpt感受态细胞中,得到zmptg菌株。
9.本技术中的实验数据证明,当同时含有2个phacab拷贝(一个在基因组上,一个在质粒上)时,phb的产量大幅度提高,为16.99%dcw。
10.进一步地,分别将基因zmo1329和zmo0367与质粒pez-pg相连构建重组质粒pez-pg
n1
和pez-pg
n2
,然后转入zmpt感受态细胞,得到菌株zmpt
n1
和zmpt
n2
。
11.本技术中分别过表达辅因子供给相关基因zwf和ppnk,结果证明,过表达zwf的效果要优于ppnk。
12.进一步地,将ada、adh2基因构建到穿梭载体pez39p中,ada、adh2基因中间用rbs序列atcacagggtctagaaggaggtcgaa连接,得到质粒pe39p-peeup,分别将质粒转入zmpt
n1
和zmpt
n2
中,得到菌株zmpt
n1-eup
和zmpt
n2-eup
。
13.进一步地,pez39p是通过将pez15a中的复制子,替换成zm4中39-032内源质粒中的复制子得到的。
14.进一步地,将ptet-phacab操纵子与pez15a的载体连接,构建重组质粒pez-pt,以pez-pt质粒为模板,pgap-cab-f和phab-r为引物,扩增phacab片段,与载体连接,构建质粒pez-pg;
15.pgap-cab-f:cttaataagttaggagaataaacatggccaccggcaaag;
16.phab-r:ggccgctactagtttaacccatatgcaagccaccattc。
17.进一步地,ada、adh2基因序列分别如seq id no.4和seq id no.5所示。
18.本发明还提供了如上述的一种生产phb的运动发酵单胞菌的构建方法在构建能够实现乙醇和phb的共同生产的工程菌中的应用。
19.本发明还提供了上述的工程菌在同时生产乙醇和phb中的应用。
20.进一步地,利用四环素诱导工程菌生产乙醇和phb。
21.综上所述,本发明的优点及积极效果为:
22.本发明构建了生产phb的运动发酵单胞菌重组菌株,并公开了其构建过程和方法。该菌株有以下的优势:
23.(1)抗噬菌体污染,运动发酵单胞菌自身有强大的限制修饰系统,较大肠杆菌等常用工程菌株有更强的抗噬菌体能力。
24.(2)降低生产成本,由于运动发酵单胞菌为兼性厌氧微生物,在发酵过程中不需要额外的溶氧控制设备,可以有效降低生产的成本。
25.(3)本研究所得的运动发酵单胞菌phb生产菌株,能够在积累phb的同时积累乙醇,并且乙醇的积累不受phb影响。
附图说明
26.图1是运动发酵单胞菌的基因工程、过程工程及连续循环发酵共产phb和乙醇的操作流程图;
27.图2是运动发酵单胞菌phb生产菌株的菌株构建流程图;
28.图3是质粒pez-pt结构图;
29.图4是样品处理方法流程图;
id no.6)。
49.本发明中,首先在zm4中建立phb合成途径,利用诱导型启动子ptet驱动phac、phaa、phab 3个基因串联,得到以上ptet-phacab操纵子,然后构建到质粒pez15a中得到质粒pez-pt。构建方法如图3。
50.以南京金斯瑞公司合成的phac、phaa、phab(保菌于通用puc质粒载体)为dna扩增模板,设计引物扩增得到目的片段(划线部分为同源臂)。
51.ptet-f:cggccgcttctagagttaagacccactttcacatttaagttg
52.ptet-r:gggagatcctttctcctctttagatc
53.ptet-cab-f:gatctcccggatccatggccaccggcaaag
54.phac-r:cttactttctctagattatggttaggctttagctttaacataacgaccag
55.phaa-f:ccataatctagagaaagtaagcacatgaccgatgttgtcattgtctctg
56.phaa-r:gtgcttactttctctagattatggttatttgcgttcaacggccaaag
57.phab-f:ctagagaaagtaagcacatgacccaacgtattgcctatc
58.phab-r:ggccgctactagtttaacccatatgcaagccaccattc
59.利用overlap pcr将ptet启动子、phac、phaa、phab三个dna片段连接成一个长片段,得到ptet-phacab操纵子。经设计,ptet启动子的3’端与phac基因的5’端拥有同源序列;phac基因的3’端与phaa基因的5’端拥有同源序列;phaa基因的3’端与phab的5’端拥有同源序列;phab基因的3’端与pez15a载体拥有同源序列。根据overlap pcr利用同源序列两两连接的原理,将ptet-phac-phaa-phab连接在一起(其中phac与phaa,phaa与phab之间的rbs序列带到phac、phaa、phab基因的同源序列中)。overlap pcr体系如下:
[0060][0061]
第一步以以上体系进行。pcr条件为98℃预变性3min;98℃变性10s,47℃退火10s,72℃延伸40s,共10个循环。第二步将第一步完成的混合物取出,加上所需引物ptet-cab-f和phab-r各1.5μl进行扩增,pcr条件为98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸40s,共26个循环。pcr完成后进行dna电泳验证,验证正确后进行dna回收得到片段phacab。
[0062]
将phacab与ptet启动子利用overlap pcr相连,pcr体系如下。
[0063][0064]
第一步以以上体系进行。pcr条件为98℃预变性3min;98℃变性10s,47℃退火10s,72℃延伸45s,共10个循环。第二步将第一步完成的混合物取出,加上所需引物ptet-f和phab-r各1.5μl进行扩增,pcr条件为98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸45s,共26个循环。pcr完成后进行dna电泳验证,验证正确后进行dna回收得到ptet-phacab操纵子。与pez15a的载体骨架通过吉布森装配的方法转到大肠杆菌dh5α细胞感受态中,pcr验证平板上的阳性克隆,过夜培养后提取其中的质粒(质粒提取按照质粒提取试剂盒标准步骤)。为提高质粒电转化效率,将得到的大肠杆菌dh5α目的菌株的质粒提取后转化到去甲基化大肠杆菌trans110细胞感受态中,pcr验证平板上的阳性克隆,过夜培养后提取其中的质粒。
[0065]
构建质粒时将获取的ptet-phacab操纵子和载体按照3:1的摩尔比例进行混合,按照下表反应体系配制完成后,在冰上静置5分钟,然后添加化学感受态,进行化学转化。利用壮观霉素抗性(100μg/ml)平板进行筛选,挑取单菌落用pez15a载体上的一对引物pez15a-f和pez5a-r对重组菌株进行菌落pcr检测,pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸80s,共30个循环,条带大小与预期一致的通过测序进行验证。
[0066][0067]
2、将重组质粒构建到目的运动发酵单胞菌中
[0068]
①
目的运动发酵单胞菌菌株感受态的制备
[0069]
用接种环挑取适量运动发酵单胞菌zm4甘油菌在rmg5固体培养基(rmg5:50g/l葡萄糖,10g/l酵母提取物,2g/l kh2po4,3g/l琼脂)平板上划线,30℃倒置培养2~3天活化;挑取活化的单菌落转接至含有10ml左右的rmg5(rmg5:50g/l葡萄糖,10g/l酵母提取物,2g/l kh2po4)液体培养基中,于30℃静置培养至对数中期用作种子液;转接种子液至含有200ml rmg5液体培养基的250ml蓝盖瓶中,控制初始od600nm数值在0.025~0.03之间。30℃静置培养至od=0.4~0.6;将装有菌液的蓝盖瓶放至冰上冷却30min后,用预冷的50ml离心管于4000rpm,离心10min收集菌体,弃上清;向离心管中加入30ml预冷的无菌水重悬洗涤菌体,混合均匀后,4000rpm离心10min弃上清;向离心管中加入30ml预冷的10%甘油重悬洗涤菌体,混合均匀后,4000rpm离心10min弃上清,并重复该步骤一次;加入1%(体积比)的预冷的
10%甘油重悬菌体,缓慢混合均匀后于冰上进行分装,每50μl分装至无菌的1.5ml离心管中,置于液氮中速冻后于-80℃保存。
[0070]
②
将重组质粒转入目的运动发酵单胞菌感受态细胞中
[0071]
取运动发酵单胞菌zm4感受态细胞于冰上,待感受态细胞融化后取50μl加入电转杯中,并在电转杯中加入1μg质粒。电转条件为1600v,25μf,200ω。电转完毕后于rmg5液体培养基中于30℃培养箱复苏。复苏4-6小时的培养物于6000rpm,1min离心,除去部分上清。悬浮菌体取100μl涂布于100μg/ml壮观霉素抗性平板,30℃培养2天。
[0072]
待有菌落生长出来之后,用pez15a载体上的一对引物pez15a-f和pez5a-r对重组菌株进行菌落pcr检测,菌落pcr的引物序列pez15a-f和pez5a-r如下:
[0073]
pez15a-f:ggcaaagccaccctatttttag
[0074]
pez5a-r:cacttcactgacaccctcat
[0075]
pcr扩增程序设置为:98℃预变性2min;98℃变性10s,55℃退火10s,72℃延伸(根据片段长度按照10s/kb进行设置),共30个循环;循环反应结束后72℃保持5min。反应体系:
[0076][0077][0078]
将获得的正确阳性克隆在所带抗性液体rmg5培养基中活化后,甘油保菌。
[0079]
3、菌株发酵测试
[0080]
将所得到的的目的菌株zm4pt在rmg5中进行发酵测试。首先取一定量甘油菌接种到含有1ml rmg5(加有1μl壮观霉素)的冻存管中,于30℃培养箱静置活化至浑浊后,倒入含有适量培养基的容器中作为发酵种子液于30℃培养箱静置培养至对数中后期,接种到rmg5以80%装瓶量于100ml三角瓶的培养基中,以不同四环素浓度(0,0.2,0.8,1.2μg/ml)进行诱导发酵。在od
600nm
处控制初始od为0.1。在发酵过程中,用紫外分光光度计测量od
600nm
处的光密度,测定不同时间点的细胞生长,并将不同时间点取得的发酵液收集,后用于hplc(高效液相色谱仪)中检测葡萄糖和乙醇的含量。采用岛津商贸有限公司agilent 1100系列高效液相色谱仪(lc-20ad);检测器为示差折光检测器(rid-10a);色谱柱为有机酸色谱柱(bio-rad aminex hpx-87h,300mm
×
7.8mm);池温度为40℃,柱温箱温度为60℃;流动相为5mm的硫酸,流速为0.5ml/min,仪器运行时初始流速设置为0.2ml/min,待柱压稳定后以0.1ml/min的流速逐渐增加至0.5ml/min;进样量为20μl。
[0081]
流动相的配置:取1.41ml色谱级浓硫酸至5l的蓝盖瓶中,用超纯水定容至5l后混合均匀,使用0.45μm孔径的水相滤膜进行过滤。将过滤后的流动相分装至1l流动相蓝盖瓶中进行超声脱气20~30min。等恢复至室温后即可使用。
[0082]
4、phb样品的处理及测定
[0083]
①
phb样品的处理
[0084]
phb定量是基于用热浓硫酸将phb聚合物转化为巴豆酸的方法。将发酵终点的培养
物用50ml离心管于冷冻离心机在25℃,4000rpm,离心8-10min收集菌体。去掉上清液,将收集完的菌体用纯水洗两次,每次洗菌同样于冷冻离心机在25℃,4000rpm,离心8-10min。将最后得到的去除上清的菌体放于65℃烘箱过夜烘干菌体。得到的干菌体研磨成粉,取20mg粉状菌体倒入25ml玻璃具塞比色管中,加入5ml浓硫酸,置于90℃下加热1h。取出冷却后在室温下,将混合物用5mm 2%硫酸稀释100倍,通过0.22μm过滤器(默克米勒孔,ma,美国)过滤,取400μl到hplc进样瓶中。样品处理方法示意如图4。
[0085]
②
phb的测定
[0086]
phb不能直接用于hplc测量,需要将其用热浓硫酸转化为巴豆酸,检测巴豆酸的含量间接得到phb的产量,由于phb标准曲线的制作也是同样的处理方法,所以此检测方法能够直接得到phb的产量。巴豆酸的测试使用aminexhpx-87h离子交换柱(bio-rad,ca,usa)进行分离,利用spd-20a,紫外检测器检测。在25℃下进行,5mm硫酸为流动相,流速为0.6ml/min。每个样品中进行了三次重复分析。
[0087]
检测不同四环素诱导浓度(0,0.2,0.8,1.2μg/ml)下的phb和乙醇产量如图5。经测试在四环素1.2μg/ml的浓度下,phb的产量为0.24%dcw。
[0088]
实施例2增加启动子强度及增加phb合成基因拷贝数
[0089]
1、替换强启动子
[0090]
1)将pez-pt质粒的诱导型启动子ptet替换为强启动子pgap得到质粒pez-pg。将pez-pg质粒转入运动发酵单胞菌zm4的感受态细胞中,得到菌株zm4pg。
[0091]
ptet启动子(seq id no.7),pgap启动子(seq id no.8)。
[0092]
以下列引物进行dna扩增:
[0093]
pgap-cab-f:cttaataagttaggagaataaacatggccaccggcaaag
[0094]
phab-r:ggccgctactagtttaacccatatgcaagccaccattc
[0095]
用pgap-cab-f和phab-r为引物,以pez-pt质粒为dna模板进行扩增,得到pgap启动子与phacab拥有同源臂的phacab片段。再用该phacab片段与pgap启动子进行overlap pcr连接得到pgap-phacab片段。将pgap-phacab片段与pez15a载体骨架(含壮观霉素筛选抗性基因)按照3:1的摩尔比例进行吉布森组装,具体比例参照下表。该体系在冰上静置5分钟,然后添加大肠杆菌dh5α感受态细胞,进行化学转化。利用壮观霉素抗性(100μg/ml)平板进行筛选,挑取单菌落,进行pcr验证,用pez15a载体上的一对引物pez15a-f和pez5a-r对单菌落进行菌落pcr检测。pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸40s,共30个循环,条带大小与预期一致的通过测序进行验证。
[0096]
[0097]
利用壮观霉素抗性(100μg/ml)平板进行筛选,挑取单菌落,进行pcr验证,用pez15a载体上的一对引物pez15a-f和pez5a-r对单菌落进行菌落pcr检测。菌落pcr的引物序列pez15a-f和pez5a-r如下:
[0098]
pez15a-f:ggcaaagccaccctatttttag
[0099]
pez5a-r:cacttcactgacaccctcat
[0100]
pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸40s,共30个循环,条带大小与预期一致的通过测序进行验证。验证得到正确的转化子命名为pez-pg。
[0101]
将pez-pg质粒电转入运动发酵单胞菌zm4的感受态细胞中,待有菌落生长出来之后,用上述pez15a载体上的一对引物pez15a-f和pez5a-r进行菌落pcr检测,pcr扩增程序设置为:98℃预变性2min;98℃变性10s,55℃退火10s,72℃延伸(根据片段长度按照10s/kb进行设置),共30个循环;循环反应结束后72℃保持5min。
[0102]
2)、菌株发酵测试
[0103]
将所得到的的目的菌株zm4pg在rmg5中进行发酵测试。首先取一定量甘油菌接种到含有1ml rmg5(加有1μl壮观霉素)的冻存管中,于30℃培养箱静置活化至浑浊后,倒入含有适量培养基的容器中作为发酵种子液于30℃培养箱静置培养至对数中后期,接种到rmg5以80%装瓶量于100ml三角瓶的培养基中进行发酵。在od
600nm
处控制初始od为0.1。在发酵过程中,用紫外分光光度计测量od值600
nm
处的光密度,测定不同时间点的细胞生长,并将不同时间点取得的发酵液收集,后用于hplc(高效液相色谱仪)中检测葡萄糖和乙醇的含量。采用岛津商贸有限公司agilent 1100系列高效液相色谱仪(lc-20ad);检测器为示差折光检测器(rid-10a);色谱柱为有机酸色谱柱(bio-rad aminex hpx-87h,300mm
×
7.8mm);池温度为40℃,柱温箱温度为60℃;流动相为5mm的硫酸,流速为0.5ml/min,仪器运行时初始流速设置为0.2ml/min,待柱压稳定后以0.1ml/min的流速逐渐增加至0.5ml/min;进样量为20μl。
[0104]
流动相的配置:取1.41ml色谱级浓硫酸至5l的蓝盖瓶中,用超纯水定容至5l后混合均匀,使用0.45μm孔径的水相滤膜进行过滤。将过滤后的流动相分装至1l流动相蓝盖瓶中进行超声脱气20~30min。等恢复至室温后即可使用。
[0105]
3)、phb样品的处理及测定
[0106]
①
phb样品的处理
[0107]
phb定量是基于用热浓硫酸将phb聚合物转化为巴豆酸的方法。将发酵终点的培养物用50ml离心管于冷冻离心机在25℃,4000rpm,离心8-10min收集菌体。去掉上清液,将收集完的菌体用纯水洗两次,每次洗菌同样于冷冻离心机在25℃,4000rpm,离心8-10min。将最后得到的去除上清的菌体放于65℃烘箱过夜烘干菌体。得到的干菌体研磨成粉,取20mg粉状菌体倒入25ml玻璃具塞比色管中,加入5ml浓硫酸,置于90℃下加热1h。取出冷却后在室温下,将混合物用5mm 2%硫酸稀释100倍,通过0.22μm过滤器(默克米勒孔,ma,美国)过滤,取400μl到hplc进样瓶中。样品处理方法示意如图4。
[0108]
②
phb的测定
[0109]
phb不能直接用于hplc测量,需要将其用热浓硫酸转化为巴豆酸,检测巴豆酸的含量间接得到phb的产量,由于phb标准曲线的制作也是同样的处理方法,所以此检测方法能
够直接得到phb的产量。巴豆酸的测试使用aminexhpx-87h离子交换柱(bio-rad,ca,usa)进行分离,利用spd-20a,紫外检测器检测。在25℃下进行,5mm硫酸为流动相,流速为0.6ml/min。每个样品中进行了三次重复分析。
[0110]
检测phb和乙醇产量如图6。
[0111]
2、将phacab片段整合到zm4基因组
[0112]
利用运动发酵单胞菌内源crispr-cas技术将ptet-phacab操纵子片段整合到运动发酵单胞菌zm4基因组上zmo0038基因的位置。整合原理如图7。
[0113]
编辑质粒所用的扩增引物如下:
[0114]
0038-gr1-f:gaaagcgtccagcaaaatacgccttctattgatgaa
[0115]
0038-gr1-r:gaacttcatcaatagaaggcgtattttgctggacgc
[0116]
0038-up-f:caccagctcaccgtctgctttttgccgacaaagcg
[0117]
0038-up-r:tcacgcccgacgccagacgggattagaaattttgtcg
[0118]
0038-up-ptet-f:ggcgtcgggcgtgattaagacccactttcacatttaagttgtttttctaatc
[0119]
phab-down-r:cgtctatctgaatatttaacgattaacccatatgcaagccacc
[0120]
0038-down-f:tcgttaaatattcagatagacggagataataaacgggagagaggtcg
[0121]
0038-down-r:gctcgagatctgatatcactcaacagatcaacc
[0122]
0038-up-pgap-f:ggcgtcgggcgtgagttcgatcaacaacccgaatcctatc
[0123]
crispr-反扩-f:gtgatatcagatctcgagctcggtacccgggtttgac
[0124]
crispr-反扩-r:gacggtgagctggtgacctgccttatctctttcccc
[0125]
测-0038-f:tgccagcttctgttggagaaaacagg
[0126]
测-0038-r:cgcaagccaagctgcgt
[0127]
菌株的稳定性是工业发酵生产应用的关键,而质粒往往容易从菌株中丢失,因此这里进一步将生产phb的途径整合在基因组。将操纵子ptet-phacab整合到zm4菌株染色体zmo0038位置(经实验室证明该位置的变化对菌株生长没有影响)。
[0128]
将空载编辑质粒pl2r(该质粒构建参考zheng et al.,nucleic acids research 47:11461-11475)在37℃处进行过夜酶切。酶切体系如下:
[0129][0130]
过夜酶切,将酶切产物进行dna回收。基因编辑工作所构建的编辑质粒,由一个间隔区间spacer引导cas蛋白靶向目的基因处进行编辑工作。该间隔区包含一个5
‘‑
ccc-3’pam位点的整个32bp序列。将间隔区设计为两条正反引物用于退火(0038-gr1-f,0038-gr1-r),将反应混合物加热到95℃,退火5min,然后逐渐冷却到室温。退火体系如下:
[0131][0132]
退火后的间隔区spacer取2μl,在18℃与线性酶切后的pl2r载体与t4连接酶进行过夜酶连,以插入将替换片段与zm4基因组连接的donor(供体dna)片段。酶连体系如下:
[0133][0134]
过夜酶连,将酶连产物进行dna回收。得到的回收产物转化到大肠杆菌dh5α菌株感受态细胞中,涂平板于lb加壮观霉素的固体培养基中,隔天挑取单菌落进行菌落pcr验证寻找正确的转化子称之为grna。验证引物为(0038-gr1-f,pez15a-r)。
[0135]
donor的设置由所替换的zmo0038上下游500bp(分别称之为up stream和down stream端),序列如下:
[0136]
0038-up stream:
[0137]
tcacgcccgacgccagacgggattagaaattttgtcgataaacgcgccgtgacgcaataattcttgtgggctggcctcgtggggtcttggttgtcttttgacctttttattacgaggtagaccggaaaccgtgttcggggcagacaagcgtccatcggatgggccgttattgtcatccaaggccataccgatttgccgtccgcctgtcagttcaacataaagctgtgccagcaattcggcatcgagcaaggcgccatgtttgacgcgatggctgcggtcaataccgtagcgggtacataatgcgtcaagggtatgtttggcaccgggatgtttgccgcgcgcaatcgccaaggtatcgaccattcgggtcatacaaatggtcggaaggccgcatttatccaattcgtaattcaaaaagccgaaatcgaaggccgcattatgggcgaccaaaggcgacaatttgataaaggaaagcagttcttgcgctttgtcggcaaaaag
[0138]
0038-down stream:
[0139]
caacagatcaaccggcaatttatccacggcatcaaattcgatcggttctttcaggcggacaaaaaagccggagacttttttcaggctgtcgagcttgccatgtggtatcgcaataccatgaccaaaacctgtcgaaccgagcgtttctctttcatgtaaccgttcggcgataaccttggggttaattagaaattctcgcccagcccattgggcaacctgctggaagagtgtttttttgttagaaggattgaaattcacctcaatcctgtccggcgcgattatatcggccagttcattcatgttttttcgcctttatatgcccacgattgcaaagatcgcagcatcagacattttctgaaagacacagcctttcagaaaataaagtcaaaattctagcaatattccttccggaaacgggggagaggggtcgataattctcttgaaaagagggagcgacctctctcccgtttattatctccgtctatctgaatatttaacga
[0140]
在其中间插上替换片段ptet-phacab,将三个片段通过overlap pcr连接成donor
(up stream-ptet cab-down stream)。然后将该donor与grna质粒的反向扩增片段(以crispr-反扩-f,crispr-反扩-r为引物进行扩增)构建成质粒。构建过程如下;
[0141]
反向扩增grna质粒,体系入下:
[0142][0143]
将得到的grna反向扩增产物按试剂盒步骤进行dna胶回收。
[0144]
吉布森组装构建质粒:
[0145]
将donor(up stream-ptet cab-down stream)与grna质粒的反向扩增片段(含壮观霉素筛选抗性基因)按照3:1的摩尔比例进行吉布森组装,具体比例参照下表。该体系在冰上静置5分钟,然后添加大肠杆菌dh5α感受态细胞,进行化学转化。利用壮观霉素抗性(100μg/ml)平板进行筛选,挑取单菌落,进行pcr验证,用pez15a载体上的一对引物pez15a-f和pez5a-r对单菌落进行菌落pcr检测。pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸20s,共30个循环,条带大小与预期一致的通过测序进行验证。
[0146][0147]
得到所得正确转化子后培养,提取所得编辑质粒。将该编辑质粒转化进zm4菌株,通过菌落pcr验证寻找到正确的转化子,以zmo0038的前后500bp外围引物(测-0038-f,测-0038-r)进行菌落pcr,若所得pcr产物通过dna凝胶电泳验证出大小为ptet-phacab将zmo0038成功替换的大小,则认为是编辑成功,将pcr原液送给公司测序。若得到已经编辑成功的菌株(ptet-phacab成功替换zm4基因组上zmo0038基因),将该菌株在rmg5培养基中传代至菌株能在rmg5生长但不能在rmg5添加壮观霉素的环境下生长,以丢掉编辑质粒。得到zmpt菌株后,在100ml三角瓶中进行80%rmg5液体培养基装瓶量的发酵,以0、0.4、0.8、1.2μg/ml四环素浓度与zm4pt对照试验,每隔3小时在超净工作台中取出1ml样品收集,测量发酵过程中的乙醇与葡萄糖的量和最后的phb产量,经测试在四环素1.2μg/ml的浓度下,zmpt菌株的phb产量为2.07%dcw。
[0148]
本实施例中利用运动发酵单胞菌内源crispr-cas技术ptet驱动的phacab片段整合到运动发酵单胞菌zm4基因组zmo0038的位点,得到菌株zmpt,发酵检测phb和乙醇产量如
图8。用同样的方法将pgap-phacab整合到运动发酵单胞菌zm4基因组zmo0038基因的位置,得到菌株zmpg。
[0149]
3、多拷贝菌株的构建
[0150]
在菌株zmpg基础上,转入pgap驱动的phacab质粒pez-pg,得到含有2个phacab拷贝的(一个在基因组zmo0038基因的位置上,一个在质粒上)菌株zmpgg。
[0151]
本实施例中将所得强启动子pgap驱动的phacab质粒pez-pg,电转入zmpt感受态细胞中,得到含有2个phacab拷贝的(一个在基因组zmo0038基因的位置上,一个在质粒上)菌株zmptg菌株,经测试,在四环素1.2μg/ml的浓度下,phb的产量为16.99%dcw。发酵检测phb和乙醇产量如图9。
[0152]
实施例3过表达辅因子供给相关基因
[0153]
由于phb积累过程中,乙酰乙酰辅酶a还原酶phab催化的步骤依赖于nadph的消耗供能。因此通过过表达运动发酵单胞菌内源的葡萄糖-6磷酸脱氢酶(zwf,zmo0367)和nad
激酶(ppnk,zmo1329)的基因来增强nadph供应,帮助提高phb产量。
[0154]
本发明先分别构建zmo1329和zmo0367与pez-pg质粒进行连接,得到重组质粒pez-pg
n1
(pgap-zmo1329-phacab)和pez-pg
n2
(pgap-zmo0367-phacab),然后转入zmpt细胞感受态得到菌株zmpt
n1
,zmpt
n2
,发酵检测phb和乙醇产量。pez-pg
n1
,pez-pg
n2
质粒构建如图10。
[0155]
相关引物如下:
[0156]
ppnk-phac-f:gttaaaaagaggagaaaggatctcccatggccaccggcaaag
[0157]
ppnk-r:tcctttctcctctttttaacataagcgaaattgttcgcg
[0158]
zwf-f:atgacaaataccgtttcgacgatg
[0159]
zwf-r:gagatcctttctcctcttttcagtcataccaagttactccatcac
[0160]
zwf-phac-f:aaagaggagaaaggatctcccatggccaccggcaaagg
[0161]
phab-r:ggccgctactagtttaacccatatgcaagccaccattc
[0162]
以ppnk-phac-f和ppnk-r为一对引物,以zwf-f和zwf-r为一对引物,分别用zm4为模板扩增50μl ppnk和zwf基因片段后进行dna回收(按试剂盒的方法进行)。然后分别用ppnk-phac-f和zwf-phac-f与phab-r扩增50μl pez-pg质粒上的phacab片段,进行dna回收。pcr体系如下。
[0163][0164][0165]
将所得ppnk和zwf片段与phacab片段进行overlap pcr连接后进行dna胶回收得到ppnk-phacab,zwf-phacab。将ppnk-phacab,zwf-phacab与反向扩增的pez-15a载体连接构建质粒pez-pg
n1
,pez-pg
n2
。将pez-pg
n1
,pez-pg
n2
分别转入zmpt感受态细胞,得到菌株zmpt
n1
,zmpt
n2
。
[0166]
本实施例中先分别构建含有zmo1329和zmo0367与pgap驱动的phacab质粒相连的
重组质粒pez-pg
n1
和pez-pg
n2
,将phacab与pez15a载体骨架分别与zmo1329和zmo0367基因进行吉布森组装构建质粒。然后转入zmpt菌株得到菌株zmpt
n1
,zmpt
n2
,发酵检测phb和乙醇产量如图11。经测试在四环素1.2μg/ml的浓度下,phb的产量分别为19.97%dcw,24.22%dcw。
[0167]
实施例4引入外源乙醇利用途径
[0168]
外源乙醇利用途径(eup)是一种由来自酿酒酵母的adh2和ada基因编码的利用乙醇的途径。它可以将1分子乙醇转化为乙酰辅酶a并产生2个nadh,而不消耗atp来提供乙酰辅酶a依赖的生物化学物质。pez39p是将pez15a中的复制子,替换成zm4中39-032内源质粒中的复制子。通过t5外切酶依赖的dna组装(teda)方法,将39-032复制子片段克隆到pez15asp载体得到pez39p。39-032复制子序列如下(seq id no.9):
[0169]
pez39p质粒的构建如下:
[0170]
①
扩增zm4菌株内源质粒p39上的39-032复制子片段
[0171]
以zm4菌株为dna扩增模板,体系如下:
[0172][0173]
将所得pcr原液进行dna溶液回收。
[0174]
②
反向扩增pez15a质粒上的载体片段
[0175]
以pez15a质粒为dna扩增模板,体系如下
[0176][0177]
将所得rcr原液进行dna胶回收。
[0178]
③
将回收后的39-032复制子片段与pez15a质粒上的载体片段通过吉布森组装构建质粒
[0179]
将39-032复制子片段与pez15a质粒上的载体片段(含壮观霉素筛选抗性基因)按照3:1的比例进行吉布森组装,具体比例参照下表。该体系在冰上静置5分钟,然后添加大肠杆菌dh5α感受态细胞,进行化学转化。利用壮观霉素抗性(100μg/ml)平板进行筛选,挑取单菌落,进行pcr验证,用pez15a载体上的一对引物39-032-seq-f和pez5a-r对单菌落进行菌落pcr检测。pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸12s,共30个循环,条带大小与预期一致的通过测序进行验证。
[0180][0181]
所用的引物序列如下:
[0182]
39-032-p-spe-f:ttccgtagtgagtactgaatctatcgaaaggcaaatttctttctcg
[0183]
39-032-p-spe-r:agaagcggccgcgaattcagtcagaaccggcgccc
[0184]
pez-dp-反扩-f:ctgaattcgcggccgc
[0185]
pez-反扩-r:attcagtactcactacggaattgc
[0186]
39-032-seq-f:cggctctaaccgaccag
[0187]
本技术中首先将eup途径的ada-adh2串联基因(以peno启动子驱动)构建到运动发酵单胞菌质粒pez39p中得到质粒pe39p-peeup。构建过程如下:
[0188]
由于构建的eup途径的质粒需转入带有壮观霉素抗性质粒的菌株中,因此需要把构建好的pez39p质粒载体中间的壮观霉素抗性基因换成卡纳霉素抗性基因。质粒pe39p-peeup的构建由pez39p质粒的反扩载体片段(抗性基因前端,抗性基因后端)、卡纳霉素抗性基因、peno-ada-adh2操纵子构成。
[0189]
①
pez39p质粒的反扩载体片段(抗性基因前端)的扩增:以pez39p质粒为dna扩增模板反向扩增
[0190][0191]
pez39p质粒的反扩载体片段(抗性基因后端)的扩增:以pez39p质粒为dna扩增模板反向扩增
[0192][0193]
卡纳霉素抗性基因的扩增:以zm4菌株为dna扩增模板
[0194][0195]
所得pcr原液均进行dna溶液回收。
[0196]
②
将得到的pez39p质粒的反扩载体片段(抗性基因前端,抗性基因后端)、卡纳霉素抗性基因三个片段通过overlap pcr连接成一个片段。经设计,pez39p质粒的反扩载体片段(抗性基因前端)的3’端与卡纳霉素基因的5’端拥有同源序列;卡纳霉素基因的3’端与pez39p质粒的反扩载体片段(抗性基因后端)的5’端拥有同源序列。根据overlap pcr利用同源序列两两连接的原理,将pez39p质粒的反扩载体片段(抗性基因前端)-卡纳霉素-pez39p质粒的反扩载体片段(抗性基因后端)连接在一起。overlap pcr体系如下:
[0197][0198]
第一步以以上体系进行。pcr条件为98℃预变性3min;98℃变性10s,47℃退火10s,72℃延伸40s,共10个循环。第二步将第一步完成的混合物取出,加上所需引物39质粒反扩-f和39p反扩后端-r各1.5μl进行扩增,pcr条件为98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸40s,共26个循环。pcr完成后进行dna电泳验证,验证正确后进行dna回收得到pez39p质粒(带卡纳霉素抗性基因)的反扩载体片段。
[0199]
③
peno-ada-adh2操纵子的构建
[0200]
ada-adh2串联基因由zm4基因组上的peno启动子驱动,首先需要以zm4菌株为dna
扩增模板扩增出peno启动子:
[0201][0202]
所得pcr原液进行dna溶液回收得到peno启动子,将peno启动子与所得ada-adh2串联基因进行overlap pcr连接成peno-ada-adh2操纵子:
[0203]
将得到的peno启动子、ada-adh2串联基因通过overlap pcr连接成一个片段。经设计,peno启动子的3’端与ada-adh2串联基因的5’端拥有同源序列。根据overlap pcr利用同源序列两两连接的原理,将peno-ada-adh2连接在一起。overlap pcr体系如下:
[0204][0205]
第一步以以上体系进行。pcr条件为98℃预变性3min;98℃变性10s,47℃退火10s,72℃延伸20s,共10个循环。第二步将第一步完成的混合物取出,加上所需引物39质粒反扩-f和39p反扩后端-r各1.5μl进行扩增,pcr条件为98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸20s,共26个循环。pcr完成后进行dna电泳验证,验证正确后进行dna回收得到peno-ada-adh2操纵子。
[0206]
③
pe39p-peeup质粒的构建
[0207]
将peno-ada-adh2操纵子片段与pez39p质粒(带卡纳霉素抗性基因)的反扩载体片段按照3:1的摩尔比例进行吉布森组装,具体比例参照下表。该体系在冰上静置5分钟,然后添加大肠杆菌dh5α感受态细胞,进行化学转化。利用卡纳霉素抗性(200μg/ml)平板进行筛选,挑取单菌落,进行pcr验证,用pez15a载体上的一对引物测39-f和pez5a-r对单菌落进行菌落pcr检测。pcr扩增程序设置为:98℃预变性3min;98℃变性10s,55℃退火10s,72℃延伸25s,共30个循环,条带大小与预期一致的通过测序进行验证。
[0208][0209][0210]
所用引物如下:
[0211]
39质粒反扩-f:cgtcccatagatctcgagc
[0212]
39质粒反扩-r:gatgtttaactcctgaattaagccgc
[0213]
39p反扩后端-f:tgtctaacaattcgttcaagccgacgc
[0214]
39p反扩后端-r:ctcgagagatctgatatcactctag
[0215]
kana-f:gcttaattcaggagttaaacatcatgagccatattcaacgggaaacg
[0216]
kana-r:ttagaaaaactcatcgagcatcaaatgaaactgc
[0217]
peno-f:tgtctatactccagttactcaatacgtaacaataatcagtttatcctaac
[0218]
peno-r:atcgaaacctttcttaaaatcttttagacgag
[0219]
测39-f:ggctctaaagaaaagacagaggc
[0220]
pez5a-r:cacttcactgacaccctcat
[0221]
然后将pe39p-peeup质粒转入zmpt
n1
和zmpt
n2
,分别得到菌株zmpt
n1-eup
和zmpt
n2-eup
。所得菌株在100ml三角瓶中以80%装瓶量,rmg5为培养基,四环素诱导剂浓度为1.2μg/ml,控制初始od
600nm
为0.1在30℃,100rpm摇床进行发酵测试。发酵检测phb和乙醇产量如图12。经测试,在该条件下zmpt
n1-eup
和zmpt
n2-eup
的phb的产量分别为26.15%dcw,37.68%dcw。
[0222]
我们将由南京金斯瑞公司合成的adh2和ada基因引入phb菌株。基因序列如下:
[0223]
ada(seq id no.4),adh2(seq id no.5)。
[0224]
将上述ada和adh2基因通过rbs序列(第1381-1406位)连接,得到如下序列片段(seq id no.10)。
[0225]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
