1.本发明涉及基于镍的负载型金属催化剂,其意在特别用于不饱和烃的加氢、更特别是用于多不饱和化合物的选择性加氢或芳族化合物的加氢。
现有技术
2.单不饱和有机化合物(例如乙烯和丙烯)是制备聚合物、塑料和具有附加值的其他化学品的基础。这些化合物由已通过蒸汽裂化或催化裂化方法处理的天然气、石脑油或瓦斯油获得。这些方法在高温下实施,并且除了所需的单不饱和化合物之外,还产生多不饱和有机化合物,例如乙炔、丙二烯和甲基乙炔(或丙炔)、1,2-丁二烯和1,3-丁二烯、乙烯基乙炔和乙基乙炔以及其他沸点相当于c5 汽油馏分(含具有5个或更多个碳原子的烃化合物的汽油)的多不饱和化合物、特别是苯乙烯类或茚类化合物。这些多不饱和化合物具有高反应性并导致聚合单元中的副反应。因此有必要在经济地利用这些馏分之前除去这些多不饱和化合物。
3.选择性加氢是为了从这些烃原料中针对性地除去不需要的多不饱和化合物而开发的主要处理方法。它可以将多不饱和化合物转化为相应的烯烃或芳族化合物,同时避免其完全饱和及由此形成相应的烷烃或环烷烃。
4.选择性加氢催化剂通常基于来自元素周期表第viii族的金属,优选钯或镍。该金属是沉积在载体上的金属颗粒的形式。金属含量、金属颗粒的尺寸和活性相在载体中的分布是对催化剂的活性和选择性产生影响的参数。
5.金属颗粒在载体中的宏观分布构成了一个重要参数,主要是在快速且连续反应的背景下,例如选择性加氢。通常希望这些元素位于载体外周的壳体中,从而避免可能导致活性缺陷和选择性损失的粒内物质转移问题。这样的催化剂也被称为“蛋壳”催化剂。
6.这样的催化剂在基于钯的选择性加氢催化剂的情况下广为人知。事实上,借助于低钯含量 (相对于催化剂而言,钯通常小于1重量%(1wt%))和合适的制备方法,可以在载体粒子外周处获得薄钯壳(fr2922784、us2010/217052)。
7.经常建议用镍来代替钯,镍是一种活性不如钯的金属,这因此需要在催化剂中加入更大量的镍。因此,相对于催化剂,基于镍的催化剂通常具有5重量%-50重量%的镍金属含量。在这些催化剂中,镍通常均匀地分布在载体内。在活性和选择性方面改善这些催化剂的一种可能途径是通过将镍以更集中的方式沉积在载体外周处的壳体上来控制镍在载体内的分布。这样的催化剂在现有技术中是已知的。
8.文献us 4 519 951描述了一种“蛋壳”催化剂,镍在多孔载体上,所述多孔载体的尺寸小于11.7nm的孔的孔体积为至少0.2ml/g,尺寸大于11.7nm的孔的孔体积为至少0.1ml/g。大于50%的镍存在于壳体中,壳体的厚度等于0.15倍的载体半径。这种催化剂用于脂肪的加氢。
9.文献cn101890351描述了一种负载型镍催化剂,其中大于90%的镍存在于700μm厚的壳体中。该催化剂是使用含氨溶液溶解镍盐制备而成。这些催化剂被用于选择性加氢应
用中。
10.文献us2012/0065442描述了一种负载型镍催化剂,其中镍微晶的尺寸分布为双峰,30%至70%的镍微晶的平均尺寸(直径)为1.0-2.5nm,其余的镍微晶的平均尺寸(直径)为3.0-4.5nm。镍既分布在厚度为直径的3%-15%的壳体上,也分布在内核处,壳体和内核的镍浓度比为3.0:1-1.3:1。至少75%的孔体积存在于尺寸大于5.0nm的孔中。
11.发明目的令人惊奇的是,本技术人已发现,通过在向包含按照非常特定的方法获得的氧化铝载体的基于镍的催化剂添加特定的有机添加剂后进行特定的水热处理,获得了催化剂,其中至少一部分的镍分布在载体外周的壳体上,另一部分镍分布在催化剂的内核中。不希望受任何理论约束,在使特定的有机添加剂与负载于特定氧化铝载体(已对该载体进行了在酸溶液的存在下的水热处理)上的基于镍的催化剂接触的步骤之后进行的水热处理,似乎使镍至少部分地从载体的内部迁移到载体的外周,从而形成镍壳。因此,本发明涉及一种新型催化剂,借助于其特定的制备方法,可以获得一种催化剂,在多不饱和化合物的选择性加氢反应或多不饱和芳族化合物的加氢反应的背景下,所述催化剂在活性和选择性方面具有至少与现有技术一样好、或甚至更好的性能品质,同时使用比现有技术中通常使用的更少量的镍相,这是因为镍活性相在载体中的更好分布,这使镍活性相更容易接近反应物,以及由于镍颗粒尺寸小于7nm,这使镍的内在活性变得更高。
12.根据本发明的第一主题涉及包含基于镍的活性相和氧化铝载体的催化剂,所述催化剂包含相对于催化剂总重量的1重量%-50重量%的元素镍,所述催化剂的特征在于:
‑ꢀ
镍既分布在载体外周的壳体上,也分布在载体的内核中,所述壳体的厚度为催化剂直径的2%-15%;
‑ꢀ
壳体和内核的镍密度比严格地大于3;
‑ꢀ
相对于催化剂中所含的镍的总重量,所述壳体包含大于25重量%的镍元素;
‑ꢀ
催化剂中以氧化物形式测量的镍颗粒的尺寸小于7nm。
13.有利地,壳体和内核的镍密度比大于或等于3.5。
14.有利地,相对于催化剂中所含的镍的总重量,所述壳体包含大于40重量%的镍元素。
15.有利地,所述催化剂的内核和壳体之间的过渡区间为催化剂直径的0.05%-3%。
16.有利地,催化剂中镍颗粒的尺寸小于5nm。
17.根据本发明的另一个主题涉及制备根据本发明的催化剂的方法,所述方法包括以下步骤:a)提供氧化铝凝胶;b)将来自步骤a)的氧化铝凝胶成型;c)对在步骤b)结束时获得的经成型的氧化铝凝胶进行热处理,所述热处理包括在酸溶液的存在下、在高压釜中、在100℃-800℃的温度下的至少一个水热处理步骤,以及在水热处理步骤之后进行的在400℃-1500℃的温度下的至少一个煅烧步骤,以获得氧化铝载体;d)进行以下步骤:d1)使氧化铝载体与至少一种含有镍活性相的至少一种前体的溶液接触;
d2)使氧化铝载体与至少一种含有至少一种有机化合物的溶液接触,所述有机化合物包含至少一个羧酸官能团、或至少一个醇官能团、或至少一个酯官能团、或至少一个酰胺官能团、或至少一个胺官能团,步骤d1)和d2)同时进行或以任何顺序单独进行;e)在低于250℃的温度下干燥在步骤d)结束时获得的催化剂前体;f)使在步骤e)结束时获得的经干燥的催化剂前体与至少一种含有至少一种有机添加剂的溶液接触,所述有机添加剂选自每分子含有1-14个碳原子的醛、每分子含有3-18个碳原子的酮或多元酮、每分子含有2-14个碳原子的醚和酯、每分子含有1-14个碳原子的醇或多元醇、以及每分子含有1-14个碳原子的羧酸或多元羧酸,有机添加剂和镍的摩尔比大于0.05mol/mol;g)在100℃-200℃的温度下、在每kg干燥气体包含5-650克水的气流下对在步骤f)结束时获得的催化剂前体进行水热处理。
18.有利地,在步骤d2)中引入的所述有机化合物与也在步骤d1)中引入的镍元素的摩尔比为0.01-5.0mol/mol。
19.有利地,步骤d1)和步骤d2)同时进行。
20.有利地,步骤d1)和/或步骤d2)通过干法浸渍进行。有利地,步骤d2)的有机化合物选自草酸、丙二酸、乙醇酸、乳酸、丙醇二酸、柠檬酸、酒石酸、丙酮酸、乙酰丙酸、乙二醇、丙-1,3-二醇、丁-1,4-二醇、甘油、木糖醇、甘露醇、山梨醇、二乙二醇、葡萄糖、γ-戊内酯、碳酸二甲酯、碳酸二乙酯、甲酰胺、n-甲基甲酰胺、乙酰胺、n-甲基乙酰胺、n,n-二甲基甲酰胺、2-吡咯烷酮、γ-内酰胺、乳酰胺、脲、丙氨酸、精氨酸、赖氨酸、脯氨酸、丝氨酸、edta。
21.有利地,步骤d2)的有机化合物不同于在步骤f)中引入的有机添加剂。
22.有利地,在步骤f)中,有机添加剂选自甲酸、甲醛、乙酸、柠檬酸、草酸、乙醇酸、丙二酸、乙醇、甲醇、甲酸乙酯、甲酸甲酯、三聚乙醛、乙醛、γ-戊内酯、葡萄糖、山梨醇和三氧杂环己烷。
23.有利地,步骤f)的有机添加剂和镍的摩尔比为0.1-5mol/mol。
24.根据本发明的另一个主题涉及将具有低于或等于300℃的终沸点的烃原料中所含的每分子含有至少2个碳原子的多不饱和化合物选择性加氢的方法,所述方法在根据本发明的催化剂的存在下、在0℃-300℃的温度、0.1-10mpa的压力下、当所述方法在液相中实施时,在0.1-10的氢气/(待加氢的多不饱和化合物)摩尔比以及0.1-200h-1
的时空速下实施,或者当所述方法在气相中实施时,在0.5-1000的氢气/(待加氢的多不饱和化合物)摩尔比以及100-40000h-1
的时空速下实施。
25.根据本发明的另一个主题涉及将具有低于或等于650℃的终沸点的烃原料中所含的至少一种芳族或多芳族化合物加氢的方法,所述方法在根据本发明的催化剂的存在下、在气相或液相中、在30℃-350℃的温度、0.1-20mpa的压力、0.1-10的氢气/(待加氢的芳族化合物)摩尔比以及0.05-50h-1
的时空速(hsv)下实施。
附图说明
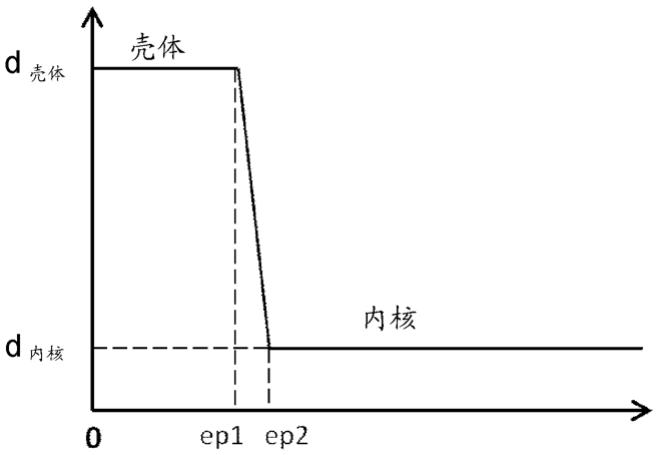
26.图1是示出了催化剂中的镍分布的图。x轴对应于催化剂的厚度,从催化剂的边缘开始测量(以
µ
m为单位)。y轴对应于镍密度(以镍的克数/mm3为单位)。镍既分布在厚度为
ep1的载体外周的壳体上,也分布在载体的内核中。壳体上的镍密度d
壳体
大于载体内核中的镍密度d
内核
。催化剂的内核和壳体之间的过渡区间的厚度表示为ep2-ep1。
27.发明详述1. 定义在下文的文字中,根据cas分类法(crc handbook of chemistry and physics,由crc press出版,主编d.r.lide,第81版,2000-2001)给出化学元素的族。例如,根据cas分类法的第viii族对应于根据新的iupac分类法的第8列、第9列和第10列的金属。
28.在本说明书中,根据iupac的惯例,“微孔”理解为是指直径小于2nm(即0.002
µ
m)的孔;“中孔”理解为是指直径大于或等于2nm(即0.002
µ
m)且小于或等于50nm(即0.05
µ
m)的孔,且“大孔”理解为是指直径大于50nm(即0.05
µ
m)的孔。
29.为了分析金属相在载体上的分布,通过castaing微探针(或电子微探针显微分析)测量壳体厚度。使用的设备是cameca xs100,其配备了可以同时分析四种元素的四个晶体单色仪。castaing微探针分析技术包括检测固体元素被高能电子束激发后由该固体发射出的x射线。为了实现这样的表征,将催化剂粒子涂覆在环氧树脂块中。将这些块抛光,直至获得通过珠粒或挤出物的直径的横截面,然后通过在金属蒸发器中沉积碳来进行金属化。电子探针沿着五个珠粒或挤出物的直径进行扫描,以获得固体的组成元素的平均分布曲线。这样的方法是本领域技术人员所熟知的,在l. sorbier等人的出版物“measurement of palladium crust thickness on catalyst by epma”(materials science and engineering 32(2012))中作了定义。它可以建立给定元素(在本文中是镍)在粒子内的分布曲线。此外,针对每次测量及由此针对每个分析步骤定义镍浓度。因此,粒子内的ni密度被定义为每mm3的ni浓度。
30.根据astm d4284-92标准,在140
°
的润湿角下,例如使用micromeritics
™
品牌的型号为autopore iii
™
的设备,通过水银测孔法测量总孔体积。
31.根据astm d3663-03标准通过氮气物理吸附法(在rouquerol f.、rouquerol j.和singh k.的著作“adsorption by powders & porous solids: principles,methodology and applications”,academic press,1999中描述的方法)测量bet比表面积。
32.中值中孔直径也被定义为这样的直径,使得在构成中孔体积的组合孔中,尺寸小于该直径的所有孔占通过采用水银测孔仪测定的总中孔体积的50%。
[0033]“镍颗粒的尺寸”被理解为意指氧化物形式的镍微晶的直径。氧化物形式的镍微晶的直径通过x射线衍射法、使用scherrer方程由位于角度2θ=43
°
(即沿晶向[200])处的衍射线的宽度来确定。该方法用于多晶样品或粉末上的x射线衍射法中,其将衍射峰的半高处的全宽与颗粒的尺寸相关联,并被详细描述在以下参考文献中:appl. cryst. (1978),11,102-113,“scherrer after sixty years: a survey and some new results in the determination of crystallite size”,j. i. langford和a. j. c. wilson。
[0034]
镍的含量通过x射线荧光法来测量。
[0035]
2. 催化剂本发明涉及包含基于镍的活性相和有利地包含硫和钠的氧化铝载体,优选由其组成的催化剂,所述催化剂包含相对于催化剂总重量的1重量%-50重量%的元素镍,所述催化剂的特征在于:
‑ꢀ
镍既分布在载体外周的壳体上,也分布在载体的内核中,所述壳体的厚度(也称为ep1)为催化剂直径的2%-15%,优选为催化剂直径的2.5%-12%,甚至更优选为催化剂直径的3%-10%,甚至更优选为催化剂直径的3%-7.5%;
‑ꢀ
壳体和内核的镍密度比(本文中也称为d
壳体
/d
内核
)严格地大于3,优选大于3.5,优选为3.8-15;
‑ꢀ
相对于催化剂中所含的镍的总重量,所述壳体包含大于25重量%、优选大于40重量%、更优选45重量%-90重量%、甚至更优选60重量%-90重量%的镍元素;
‑ꢀ
催化剂中以氧化物形式测量的镍颗粒的尺寸小于7nm,优选小于5nm,更优选小于4nm,甚至更优选小于3nm。
[0036]
有利地,催化剂的内核和壳体之间的过渡区间(这里也称为内核/壳体过渡区间,或根据图1中的符号表示为ep2-ep1),与沿着从催化剂的边缘到催化剂中心的催化剂厚度测量的镍密度的变化有关,是非常急变的。优选地,内核/壳体过渡区间为催化剂直径的0.05%-3%,优选为催化剂直径的0.5%-2.5%。
[0037]
相对于催化剂的总重量,根据本发明的所述催化剂中的镍含量有利地为1重量%-50重量%,更优选为2重量%-40重量%,甚至更优选为3重量%-35重量%,甚至更优选为5重量%-25重量%。“重量%”值基于镍的元素形式。
[0038]
根据本发明的催化剂可以被描述为“半蛋壳”催化剂,其中镍在载体外周处的浓度高于其在载体内核中的浓度,所述载体的内核中的镍浓度不为零。
[0039]
催化剂的比表面积通常为10m2/g-200m2/g,优选为25m2/g-110m2/g,更优选为40m2/g-100m2/g。
[0040]
催化剂的总孔体积通常为0.1ml/g-1ml/g,优选为0.2ml/g-0.8ml/g,特别优选为0.3ml/g-0.7ml/g。
[0041]
催化剂的活性相不含来自第vib族的金属。特别地,它不含钼或钨。
[0042]
所述催化剂(以及用于制备催化剂的载体)为粒子形式,其直径有利地为0.5-10mm。粒子可以具有本领域技术人员已知的任何形式,例如珠粒(优选直径为1-8mm)、挤出物、片或空心圆柱体的形式。优选地,催化剂(以及用于制备催化剂的载体)为挤出物形式,其直径为0.5-10mm,优选为0.8-3.2mm,非常优选为1.0-2.5mm,长度为0.5-20mm。挤出物的“直径”是指在这些挤出物的横截面中围成的圆的直径。催化剂可以有利地以圆柱形、多叶形、三叶形或四叶形挤出物的形式呈现。优选地,其形状为三叶形或四叶形。叶片的形状可以根据现有技术中所有已知的方法来调整。
[0043]
3. 载体本节中提到的氧化铝的特性相当于镍活性相浸渍之前的氧化铝(即在制备根据本发明的催化剂的方法的步骤c)结束时获得的氧化铝载体)的特性。
[0044]
根据本发明,载体是氧化铝,也就是说,相对于载体的重量,载体包含至少95重量%、优选至少98重量%、特别优选至少99重量%的氧化铝。所述氧化铝通常具有δ、γ或θ氧化铝类型的晶体结构,单独地或作为混合物。
[0045]
根据本发明,氧化铝载体可以包含杂质,例如来自根据cas分类法第iia族、第iiib族、第ivb族、第iib族、第iiia族、第iva族的金属的氧化物,优选是二氧化硅、二氧化钛、二氧化锆、氧化锌、氧化镁和氧化钙,或者是碱金属,优选是锂、钠或钾,和/或碱土金属,优选
是镁、钙、锶或钡,或者是硫。
[0046]
有利地,相对于氧化铝载体的总重量,所述氧化铝载体的硫含量为0.001重量%-2重量%,相对于所述氧化铝凝胶的总重量,所述氧化铝载体的钠含量为0.001重量%-2重量%。
[0047]
氧化铝的比表面积通常为10m2/g-250m2/g,优选为30m2/g-200m2/g,更优选为50m2/g-150m2/g。
[0048]
氧化铝的孔体积通常为0.1ml/g-1.2ml/g,优选为0.3ml/g-0.9ml/g,特别优选为0.5ml/g-0.9ml/g。
[0049]
制备催化剂的方法根据本发明的另一个主题涉及制备根据本发明的催化剂的方法,所述方法包括至少以下步骤:a)提供氧化铝凝胶;b)将来自步骤a)的氧化铝凝胶成型;c)对在步骤b)结束时获得的经成型的氧化铝凝胶进行热处理,所述热处理包括在酸溶液的存在下、在高压釜中、在100℃-800℃的温度下的至少一个水热处理步骤,以及在水热处理步骤之后进行的在400℃-1500℃的温度下的至少一个煅烧步骤,以获得氧化铝载体;d)进行一系列以下步骤:d1)使氧化铝载体与至少一种含有镍活性相的至少一种前体的溶液接触;d2)使氧化铝载体与至少一种含有至少一种有机化合物的溶液接触,所述有机化合物包含至少一个羧酸官能团、或至少一个醇官能团、或至少一个酯官能团、或至少一个酰胺官能团、或至少一个胺官能团,步骤d1)和d2)同时进行或以任何顺序单独进行;e)在低于250℃的温度下干燥在步骤d)结束时获得的催化剂前体;e1)任选地,在250℃-1000℃的温度下对在步骤e)结束时获得的经干燥的催化剂前体进行热处理以获得经煅烧的催化剂前体;f)使在步骤e)结束时获得的经干燥的催化剂前体,任选地在步骤e1)结束时获得的经煅烧的催化剂前体与至少一种含有至少一种有机添加剂的溶液接触,所述有机添加剂选自每分子含有1-14个碳原子的醛、每分子含有3-18个碳原子的酮或多元酮、每分子含有2-14个碳原子的醚或酯、每分子含有1-14个碳原子的醇或多元醇、以及每分子含有1-14个碳原子的羧酸或多元羧酸,有机添加剂和镍的摩尔比大于0.05mol/mol;g)在100℃-200℃的温度下、在每kg干燥气体包含5-650克水的气流下对在步骤f)结束时获得的催化剂前体进行水热处理;h)任选地,在50℃-200℃下,在每千克干燥气体包含严格小于5克水的水含量的气流下对在步骤g)结束时获得的催化剂前体进行干燥的步骤。
[0050]
除了步骤d1)和d2)之外,步骤a)至h)的顺序不可改变。可以插入中间步骤(特别是额外的干燥步骤),并且某些步骤(例如步骤d1)和/或d2))可以连续地进行若干次。最后,在步骤h)结束时,在使用催化剂之前可以增加额外的步骤。
[0051]
优选地,在成型步骤b)结束时(但在进行步骤c)之前)进行干燥步骤,然后进行煅烧步骤。
[0052]
优选地,步骤e1)和步骤h)不是任选的。
[0053]
下面详细描述所述制备方法的步骤a)至步骤h)。
[0054]
步骤a)根据本发明的催化剂包含由氧化铝凝胶获得的氧化铝载体,所述凝胶主要包含水合氧化铝(aluminum oxy(hydroxide) )(alo(oh))类型的前体—也被称为勃姆石。
[0055]
根据本发明,氧化铝凝胶(或另称为勃姆石凝胶)是通过改变ph诱导铝盐的碱性和/或酸性溶液的沉淀或本领域技术人员已知的任何其他方法合成的(p.euzen, p. raybaud, x. krokidis, h. toulhoat, j.l. le loarer, j.p. jolivet和c. froidefond, alumina,
ꢀ“
handbook of porous solids”, 由f. sch
ü
th, k.s.w. sing和j. weitkamp编辑,wiley-vch, weinheim, germany, 2002, 第1591-1677页)。
[0056]
通常,沉淀反应在5℃-80℃的温度、在6-10的ph下进行。优选地,温度为35℃-70℃,ph为6-10。
[0057]
根据一个实施方案,氧化铝凝胶通过使酸性铝盐的水溶液与碱性溶液接触而获得。例如,所述酸性铝盐选自硫酸铝、硝酸铝或氯化铝,优选地,所述酸性盐为硫酸铝。所述碱性溶液优选选自氢氧化钠或氢氧化钾。
[0058]
替代地,可以使碱性铝盐溶液与酸溶液接触,所述碱性铝盐可以选自铝酸钠和铝酸钾。在一个非常优选的变型中,所述凝胶通过使铝酸钠溶液与硝酸接触而获得。铝酸钠溶液的浓度有利地为10-5
至10-1
mol
·
l-1
,优选地,所述浓度为10-4
至10-2
mol
·
l-1
。
[0059]
根据另一个实施方案,氧化铝凝胶通过使酸性铝盐的水溶液与碱性铝盐溶液接触而获得。
[0060]
步骤b)载体可以有利地通过本领域技术人员已知的任何技术来成型。例如,可以通过捏合-挤出、通过造粒、通过滴凝(油滴)法、通过在旋转板上造粒或通过本领域技术人员熟知的任何其他方法进行成型。根据本发明的催化剂可以任选地以挤出物、片、珠粒的形式制造和使用。根据本发明,有利的成型方法是挤出,优选的挤出物形状是圆柱形、扭曲的圆柱形或多叶形(例如2叶、3叶、4叶或5叶)。
[0061]
在一个特定的实施方案中,对在步骤a)结束时获得的氧化铝凝胶实施捏合的步骤,优选在酸性介质中。所用的酸例如可以是硝酸。该步骤通过已知的设备进行,例如能够将凝胶转化为具有糊料的稠度的产物的z型臂混合机、研磨式混合机、连续式单螺杆或双螺杆。根据一个有利的实施方案,将一种或多种被称为“成孔剂”的化合物引入捏合介质中。这些化合物具有在加热时降解并因此在载体中产生孔隙的特性。例如,木粉、木炭、焦油和塑料可以被用作成孔化合物。在捏合之后,使由此获得的糊料通过挤出模头。通常,挤出物的直径为0.5-10mm,优选0.8-3.2mm,非常优选1.0-2.5mm,长度为0.5-20mm。这些挤出物可以是圆柱形、多叶形(例如三叶形或四叶形)。
[0062]
在将载体成型之后,在根据所述方法的步骤c)进行水热处理之前,任选地干燥所述载体。例如,所述干燥在50℃-200℃的温度下进行。在根据所述方法的步骤c)进行水热处理之前,任选地煅烧经干燥的载体。例如,所述煅烧在200℃-1000℃的温度下,在存在或不存在每千克干燥空气包含高达150克水的气流的情况下进行。
[0063]
步骤c)
然后,对在步骤b)结束时获得的载体实施热处理步骤,这使其具有满足所设想的应用的物理性质。
[0064]
术语“水热处理”是指在高于室温的温度下、在水的存在下通过高压釜的处理。
[0065]
在该水热处理过程中,可以以不同的方式处理经成型的氧化铝。因此,在通过高压釜之前,可以用酸溶液浸渍所述氧化铝,氧化铝的水热处理可以在气相或液相中进行,高压釜的这种气相或液相可以是酸性的或非酸性的。在进行水热处理之前,所述浸渍可以干式进行,或通过将氧化铝浸在酸性水溶液中进行。术语“干法浸渍”是指将氧化铝与体积小于或等于经处理的氧化铝的总孔体积的溶液接触。优选地,浸渍是干式进行的。
[0066]
也可以对未经事先用酸性溶液浸渍的挤出的载体进行处理,在这种情况下,酸度由高压釜的水溶液提供。
[0067]
酸性水溶液包含至少一种用于溶解挤出物中的至少一部分氧化铝的酸性化合物。术语“用于溶解挤出物中的至少一部分氧化铝的酸性化合物”被理解为意指任何与氧化铝挤出物接触后溶解至少一部分铝离子的酸性化合物。该酸应优选溶解氧化铝挤出物中的至少0.5重量%的氧化铝。
[0068]
优选地,这种酸选自强酸,例如硝酸、盐酸、高氯酸、硫酸或在使其水溶液的ph小于4的浓度下使用的弱酸(例如乙酸),或这些酸的混合物。
[0069]
根据一个优选的实施方案,水热处理在硝酸和乙酸(单独地或作为混合物)的存在下进行。高压釜优选是旋转篮式高压釜,例如专利申请ep-a-0 387 109中定义的那种。
[0070]
水热处理也可以在饱和蒸气压或在至少等于与处理温度对应的饱和蒸气压的70%的水蒸气分压下进行。
[0071]
优选地,水热处理在100℃-800℃,优选200℃-700℃的温度下进行通常30分钟至8小时,优选30分钟至3小时。
[0072]
优选地,在高压釜中进行水热处理之后进行的煅烧步骤在通常为400℃-1500℃、优选800℃-1300℃的温度下在空气中进行通常1至5小时,所述空气的水含量通常为每千克干燥空气0-700克水。
[0073]
在步骤c)结束时,所获得的氧化铝表现出如上所述的特定质构性质。
[0074]
步骤d)步骤d1)按照步骤d1)的实施,可根据本领域技术人员熟知的方法,通过干法浸渍或过量浸渍,或通过沉积-沉淀,使载体与含有镍前体的溶液接触。
[0075]
所述步骤d1)优选通过浸渍所述载体来进行,这包括例如使载体与至少一种含有镍前体的水溶液接触。所述溶液的ph可以通过任选添加酸或碱来改变。
[0076]
优选地,所述步骤d1)通过干法浸渍来进行,这包括使载体与至少一种含有至少一种镍前体、优选由其组成的溶液接触,该溶液的体积是待浸渍的载体的孔体积的0.25-1.5倍。
[0077]
优选地,所述镍前体以水溶液的形式引入,例如以硝酸盐、碳酸盐、乙酸盐、氯化物或草酸盐的形式,以多元酸或酸醇及其盐形成的配合物的形式,以与乙酰丙酮化物形成的配合物的形式,或以任何其他可溶于水溶液的无机衍生物的形式,使所述镍前体与所述载体接触。优选地,有利地使用硝酸镍、氯化镍、乙酸镍或碱式碳酸镍作为镍前体。非常优选
地,所述镍前体是硝酸镍。
[0078]
根据另一种变型,所述水溶液可以含有氨水或nh
4
铵离子。
[0079]
溶液中的镍浓度根据浸渍类型(干法浸渍或过量浸渍)和载体的孔体积进行调整,从而获得:对于负载型催化剂来说,相对于催化剂的总重量的镍含量为1重量%-50重量%、更优选2重量%-40重量%、甚至更优选3重量%-35重量%、甚至更优选5重量%-25重量%的镍元素。
[0080]
步骤d2)按照所述步骤d2)的实施,可通过本领域技术人员熟知的任何方法使所述载体与至少一种含有至少一种有机化合物的溶液接触,所述有机化合物包含至少一个羧酸官能团、或至少一个醇官能团、或至少一个酯官能团、或至少一个酰胺官能团、或至少一个胺官能团。这是因为另外注意到,在有机化合物(上文所述)的存在下制备的根据本发明的催化剂比在没有这种类型的有机化合物的情况下制备的催化剂更有活性。这种效果与镍颗粒的尺寸减小有关。
[0081]
特别地,所述步骤d2)可根据本领域技术人员熟知的方法通过干法浸渍或过量浸渍来进行。优选地,所述步骤d2)通过干法浸渍进行,这包括使催化剂的载体与体积为待浸渍的载体的孔体积的0.25-1.5倍的所述溶液接触。
[0082]
含有至少一种有机化合物的所述溶液(所述有机化合物包含至少一个羧酸官能团、或至少一个醇官能团、或至少一个酯官能团、或至少一个酰胺官能团、或至少一个胺官能团)可以是水溶液或有机溶液(例如甲醇或乙醇或苯酚或丙酮或甲苯或二甲亚砜(dmso))或由水和至少一种有机溶剂的混合物组成。所述有机化合物事先至少部分地以所需浓度溶解在所述溶液中。优选地,所述溶液是水溶液或含有乙醇。更优选地,所述溶液是水溶液。所述溶液的ph可以通过任选加入酸或碱来改变。在另一个可能的实施方案中,所述溶剂可以不存在于浸渍溶液中。
[0083]
在其中步骤d2)通过干法浸渍或过量浸渍(优选干法浸渍)进行的实施方案中,采用至少一种含有至少所述有机化合物的溶液浸渍载体可以有利地通过至少两个浸渍周期实施,每个周期中使用相同或不同的有机化合物。在这种情况下,有利地在每次浸渍之后实施干燥和任选的热处理。
[0084]
有利地,在步骤d2)中引入的所述有机化合物与也在步骤d1)中引入的镍元素的摩尔比为0.01-5.0mol/mol,优选0.05-2.0mol/mol,更优选0.1-1.5mol/mol,仍更优选0.3-1.2mol/mol。
[0085]
根据步骤d2)的有机化合物可以在同一分子中包含若干个相同或不同的羧酸、醇、酯、酰胺或胺有机官能团。根据步骤d2)的有机化合物可以包含选自羧酸、醇、酯、酰胺或胺有机官能团的若干种有机官能团的组合。
[0086]
a)包含至少一个羧酸官能团的有机化合物在根据本发明的一个实施方案中,所述有机化合物包含至少一个羧酸官能团。
[0087]
包含至少一个羧酸官能团的所述有机化合物可以是饱和或不饱和脂族有机化合物或芳族有机化合物。优选地,饱和或不饱和脂族有机化合物包含1-9个碳原子,优选2-7个碳原子。优选地,所述芳族有机化合物包含7-10个碳原子,优选7-9个碳原子。
[0088]
包含至少一个羧酸官能团的所述饱和或不饱和脂族有机化合物或所述芳族有机化合物可选自一元羧酸、二元羧酸、三元羧酸或四元羧酸。
内酰胺、己内酰胺、乙酰亮氨酸、n-乙酰天冬氨酸、氨基马尿酸、n-乙酰谷氨酸、4-乙酰氨基苯甲酸、乳酰胺和羟基乙酰胺、脲、n-甲基脲、n,n
′‑
二甲基脲、1,1-二甲基脲和四甲基脲,以其任何一种异构形式。
[0103]
e)包含至少一个胺官能团的有机化合物在根据本发明的另一个实施方案中,所述有机化合物包含至少一个胺官能团。所述有机化合物包含1-20个碳原子,优选1-14个碳原子,仍更优选2-8个碳原子。
[0104]
在根据本发明的一个实施方案中,所述有机化合物包含至少一个胺官能团,对应于经验式c
x
nyhz,其中1≤x≤20,1≤y≤x,2≤z≤2x 2。更具体地说,所述有机化合物选自乙二胺、二氨基己烷、四亚甲基二胺、六亚甲基二胺、四甲基乙二胺、四乙基乙二胺、二亚乙基三胺和三亚乙基四胺。
[0105]
在根据本发明的一个实施方案中,所述有机化合物包含至少一个胺官能团和至少一个羧酸官能团(氨基酸)。当所述化合物为氨基酸时,其优选选自丙氨酸、精氨酸、赖氨酸、脯氨酸、丝氨酸、苏氨酸或乙二胺四乙酸(edta)。
[0106]
在所有上述实施方案中,所述有机化合物选自草酸、丙二酸、乙醇酸、乳酸、丙醇二酸、柠檬酸、酒石酸、丙酮酸、乙酰丙酸、乙二醇、丙-1,3-二醇、丁-1,4-二醇、甘油、木糖醇、甘露醇、山梨醇、二乙二醇、葡萄糖、γ-戊内酯、碳酸二甲酯、碳酸二乙酯、甲酰胺、n-甲基甲酰胺、乙酰胺、n-甲基乙酰胺、n,n-二甲基甲酰胺、2-吡咯烷酮、γ-内酰胺、乳酰胺、脲、丙氨酸、精氨酸、赖氨酸、脯氨酸、丝氨酸、edta。
[0107]
浸渍步骤d1)和d2)镍催化剂的制备方法包括若干种实施方案。它们的不同之处特别在于引入有机化合物和镍前体的顺序。可以在镍前体已与载体接触之后,或在镍前体与载体接触之前,或在镍与载体接触的同时使有机化合物与载体接触。
[0108]
第一实施方案包括在所述步骤d2)之前进行所述步骤d1)。
[0109]
第二实施方案包括在所述步骤d1)之前进行所述步骤d2)。
[0110]
任选地在镍前体和/或有机化合物的存在下,将使载体与镍前体接触(步骤d1))和使载体与至少一种含有至少一种有机化合物的溶液接触(步骤d2))的每个步骤d1)和d2)进行至少一次,并可有利地进行多次,所述有机化合物包含至少一个羧酸官能团、或至少一个醇官能团、或至少一个酯官能团、或至少一个酰胺官能团、或至少一个胺官能团,在每个步骤d1)和/或步骤d2)中的所述有机化合物各自相同或不同,步骤d1)和d2)的所有可能的实施组合包括在本发明的范围内。
[0111]
第三实施方案包括同时进行所述步骤d1)和所述步骤d2)(共接触)。该实施方案可有利地包括实施一个或多个步骤d1),任选地在每个步骤d1)中使用相同或不同的镍前体。特别地,一个或多个步骤d1)在所述共接触步骤之前和/或有利地在其之后进行,任选地在每个步骤中使用相同或不同的镍前体。该实施方案可包括若干个共接触步骤:任选地在镍前体和/或有机化合物的存在下,将步骤d1)和步骤d2)同时进行若干次,在每个共接触步骤中的所述有机化合物相同或不同。
[0112]
优选地,在每个接触步骤之后可以进行中间干燥步骤。中间干燥步骤在低于250℃、优选15-240℃、更优选30-220℃、仍更优选50-200℃、甚至更优选70-180℃的温度下,在每千克干燥气体包含小于150克水,优选每千克干燥气体包含小于50克水的气流下进行通
常10分钟至24小时。有利地,当进行中间干燥步骤时,可以进行中间煅烧步骤。中间煅烧步骤在250-1000℃、优选250-750℃的温度下,在每千克干燥气体包含小于150克水,优选每千克干燥气体包含小于50克水的气流下进行通常15分钟至10小时。所述气体可以包含氧气、氮气或惰性气体,优选该气体是空气。
[0113]
有利地,在每个接触步骤之后,无论其是使镍前体与载体接触的步骤,还是使有机化合物与载体接触的步骤,还是使镍前体和有机化合物同时与载体接触的步骤,可以让经浸渍的载体熟化,其任选地在中间干燥步骤之前进行。熟化可以使溶液均匀地分布在载体中。当进行熟化步骤时,所述步骤有利地在大气压或减压下、在惰性气氛或含氧气氛或含水气氛下、在10℃-50℃、优选在环境温度下进行。通常,熟化时长小于四十八小时,优选地,五分钟至五小时是足够的。不排除更长的时间,但不一定提供任何改善。
[0114]
步骤e)干燥步骤在每千克干燥气体包含小于150克水、优选每千克干燥气体包含小于50克水的气流下、在低于250℃、优选15℃-240℃、更优选30℃-220℃、仍更优选50℃-200℃、甚至更优选70℃-180℃的温度下进行10分钟至24小时。不排除更长的时间,但不一定提供任何改善。
[0115]
所述气体可以包含氧气、氮气或惰性气体,优选该气体是空气。
[0116]
任选的步骤e1)任选的煅烧步骤在每千克干燥气体包含小于150克水、优选每千克干燥气体包含小于50克水的气流下、在250℃-1000℃、优选250℃-750℃的温度下进行。这种热处理的时长通常为15分钟至10小时。不排除更长的时间,但不一定提供任何改善。
[0117]
该气体可以包含氧气、氮气或惰性气体,优选该气体是空气。
[0118]
在步骤e)或步骤e1)结束时,镍被均匀地分布在载体上。
[0119]
步骤f)根据所述制备催化剂的方法的步骤f),使在步骤e)结束时(任选地在步骤e1)结束时)获得的催化剂前体与至少一种包含至少一种有机添加剂的溶液接触,所述添加剂选自每分子含有1-14(优选2-12)个碳原子的醛;每分子含有3-18(优选3-12)个碳原子的酮或多元酮;每分子含有2-14(优选3-12)个碳原子的醚或酯;每分子含有1-14(优选2-12)个碳原子的醇或多元醇;每分子含有1-14(优选1-12)个碳原子的羧酸或多元羧酸。有机添加剂可以由上述各种官能团的组合组成。
[0120]
优选地,所述有机添加剂选自甲酸hcooh、甲醛ch2o、乙酸ch3cooh、柠檬酸、草酸、乙醇酸(hooc-ch
2-oh)、丙二酸(hooc-ch
2-cooh)、乙醇、甲醇、甲酸乙酯hcooc2h5、甲酸甲酯hcooch3、三聚乙醛(ch
3-cho)3、乙醛c2h4o、γ-戊内酯(c5h8o2)、葡萄糖、山梨醇和三氧杂环己烷。
[0121]
特别优选地,所述有机添加剂是甲酸。
[0122]
至关重要的是,向催化剂添加有机添加剂的步骤(步骤f))是在使载体与镍活性相的前体接触的步骤之后进行的。
[0123]
优选地,所述步骤f)通过用包含至少一种上述有机添加剂的溶液浸渍在步骤e)或步骤e1)的实施结束时获得的催化剂前体来进行。浸渍通常在水溶液中、或有机溶液中、或在水溶液或有机溶液的悬浮液中进行,优选是在水溶液中进行。当在有机溶液或悬浮液中
进行操作时,优选使用醇或多元醇、二醇或聚二醇作为有机溶剂。
[0124]
优选地,所述步骤f)通过干法浸渍进行,这包括使在步骤e)或步骤e1)的实施结束时获得的催化剂前体与包含至少一种上述有机添加剂的溶液接触,该溶液的体积为待浸渍的催化剂前体的孔体积的0.25-1.5倍。
[0125]
浸渍通常在0℃-50℃、优选10℃-40℃的温度、特别优选在室温下进行。
[0126]
根据本发明,有机添加剂和镍的摩尔比大于0.05mol/mol,优选为0.1-5mol/mol,更优选为0.12-3mol/mol,甚至更优选为0.15-2.5mol/mol。
[0127]
步骤g)按照根据本发明的制备催化剂的方法的步骤g),在100℃-200℃、优选130℃-170℃、更优选约150℃的温度、在每千克干燥气体包含5-650克水、优选每千克干燥气体包含7-150克水、甚至更优选每千克干燥气体包含10-50克水的气流下对由步骤f)产生的产物进行水热处理。所述气体可以包含氧气、氮气或惰性气体,优选地,所述气体为空气。
[0128]
水热处理的时长通常为30分钟至5小时,优选1至3小时。
[0129]
步骤h(任选的)在步骤g)之后,可以在每千克干燥气体中的水含量严格地小于5克水的气流下、在50℃-200℃的温度下进行干燥步骤h),有利地,其时长为30分钟至5小时,优选1至3小时。
[0130]
所述气体可以包含氧气、氮气或惰性气体,优选地,所述气体为空气。
[0131]
在步骤g)或任选的步骤h)结束时,获得如图1所示的“半蛋壳”催化剂,其特征已在上文中描述。
[0132]
在根据本发明的一个实施方案中,催化剂的制备非原位进行,即在将催化剂装入用于选择性加氢或芳族化合物的加氢的方法的反应单元之前进行。
[0133]
步骤i)(任选的)在催化反应器中使用催化剂和实施加氢方法之前,在步骤g)或h)之后,在还原性气体的存在下有利地进行至少一个还原处理步骤i),从而获得包含至少部分地金属形式的镍的催化剂。
[0134]
这种处理可以活化所述催化剂并形成金属颗粒,特别是零价态的镍。所述还原处理可以原位或非原位进行,也就是说,在将催化剂装入加氢反应器之后或之前进行。
[0135]
还原性气体优选是氢气。氢气可以以纯的或作为混合物(例如,氢气/氮气、氢气/氩气、或氢气/甲烷混合物)使用。在氢气作为混合物使用的情况下,可以设想所有比例。
[0136]
所述还原处理在120℃-500℃、优选150℃-450℃的温度下进行。当催化剂不经历钝化或在钝化前经历还原处理时,还原处理在180℃-500℃、优选200℃-450℃、更优选350℃-450℃的温度下进行。当催化剂预先经历钝化时,还原处理通常在120℃-350℃、优选150℃-350℃的温度下进行。
[0137]
还原处理的时长通常为2-40小时,优选3-30小时。将温度升高至所需的还原温度通常是缓慢的,例如设定为0.1-10℃/分钟,优选0.3-7℃/分钟。
[0138]
以升/小时/克催化剂表示的氢气流量为0.01-100升/小时/克催化剂,优选为0.05-10升/小时/克催化剂,仍更优选为0.1-5升/小时/克催化剂。
[0139]
选择性加氢方法本发明的另一个主题是将终沸点低于或等于300℃的烃原料中包含的每分子含有
至少2个碳原子的多不饱和化合物,例如二烯烃和/或炔类和/或烯基芳族化合物(也称为苯乙烯类)选择性加氢的方法,所述方法在通过上文描述的制备方法获得的催化剂的存在下、在0℃-300℃的温度、0.1-10mpa的压力下,当所述方法在液相中进行时,在氢气/(待加氢的多不饱和化合物)摩尔比为0.1-10,时空速为0.1-200h-1
下进行,或当所述方法在气相中进行时,在氢气/(待加氢的多不饱和化合物)摩尔比为0.5-1000,时空速为100-40000h-1
下进行。
[0140]
单不饱和有机化合物(例如乙烯和丙烯)是制备聚合物、塑料和其他具有附加值的化学品的基础。这些化合物由已通过蒸汽裂化或催化裂化方法处理的天然气、石脑油或瓦斯油获得。这些方法在高温下实施,并且除了所需的单不饱和化合物之外,还产生多不饱和有机化合物,例如乙炔、丙二烯和甲基乙炔(或丙炔)、1,2-丁二烯和1,3-丁二烯、乙烯基乙炔和乙基乙炔以及其他沸点相当于c5 馏分(具有至少5个碳原子的烃类化合物)的多不饱和化合物、特别是二烯属或苯乙烯类或茚类化合物。这些多不饱和化合物具有高反应性并导致聚合单元中的副反应。因此有必要在经济地利用这些馏分之前除去这些多不饱和化合物。
[0141]
选择性加氢是为了从这些烃原料中针对性地除去不期望的多不饱和化合物而开发的主要处理方法。它能够将多不饱和化合物转化为相应的烯烃或芳族化合物,同时避免其完全饱和,并因此形成相应的烷烃或环烷烃。在蒸汽裂化汽油用作原料的情况下,选择性加氢还可以将烯基芳族化合物选择性加氢以产生芳族化合物,同时避免芳环的加氢。
[0142]
在选择性加氢方法中处理的烃原料具有低于或等于300℃的终沸点并且每分子含有至少2个碳原子并且包含至少一种多不饱和化合物。术语“多不饱和化合物”旨在表示包含至少一个炔属官能团和/或至少一个二烯官能团和/或至少一个烯基芳族官能团的化合物。
[0143]
更具体地,所述原料选自蒸汽裂化c2馏分、蒸汽裂化c2-c3馏分、蒸汽裂化c3馏分、蒸汽裂化c4馏分、蒸汽裂化c5馏分和蒸汽裂化汽油(也称为裂解汽油或c5 馏分)。
[0144]
有利地用于实施根据本发明的选择性加氢方法的蒸汽裂化c2馏分具有例如以下组成:40重量%-95重量%的乙烯,约0.1重量%-5重量%的乙炔,余量基本上是乙烷和甲烷。在一些蒸汽裂化c2馏分中,还可能存在0.1重量%-1重量%的c3化合物。
[0145]
有利地用于实施根据本发明的选择性加氢方法的蒸汽裂化c3馏分具有例如以下平均组成:约90重量%的丙烯,约1重量%-8重量%的丙二烯和甲基乙炔,余量基本上是丙烷。在一些c3馏分中,还可能存在0.1重量%-2重量%的c2化合物和c4化合物。
[0146]
c2-c3馏分也可有利地用于实施根据本发明的选择性加氢方法。它具有例如以下组成:约0.1重量%-5重量%的乙炔,约0.1重量%-3重量%的丙二烯和甲基乙炔,约30重量%的乙烯,约5重量%的丙烯,余量基本上是甲烷、乙烷和丙烷。该原料还可能含有0.1重量%-2重量%的c4化合物。
[0147]
有利地用于实施根据本发明的选择性加氢方法的蒸汽裂化c4馏分具有例如以下按重量计的平均组成:1重量%的丁烷、46.5重量%的丁烯、51重量%的丁二烯、1.3重量%的乙烯基乙炔和0.2重量%的丁炔。在一些c4馏分中,还可能存在0.1重量%-2重量%的c3化合物和c5化合物。
[0148]
有利地用于实施根据本发明的选择性加氢方法的蒸汽裂化c5馏分具有例如以下
组成:21重量%的戊烷、45重量%的戊烯和34重量%的戊二烯。
[0149]
有利地用于实施根据本发明的选择性加氢方法的蒸汽裂化汽油或裂解汽油相当于具有通常为0-300℃、优选10-250℃的沸点的烃馏分。存在于所述蒸汽裂化汽油中的待加氢的多不饱和烃特别是二烯烃化合物(丁二烯、异戊二烯、环戊二烯等)、苯乙烯类化合物(苯乙烯、α-甲基苯乙烯等)和茚类化合物(茚等)。蒸汽裂化汽油通常包含c5-c12馏分以及痕量的c3、c4、c13、c14、c15(例如,对于这些馏分中的每一种而言,其为0.1重量%-3重量%)。例如,由裂解汽油形成的原料通常具有如下组成:5-30重量%的饱和化合物(链烷烃和环烷烃),40重量%-80重量%的芳族化合物,5重量%-20重量%的单烯烃,5重量%-40重量%的二烯烃,1重量%-20重量%的烯基芳族化合物,这些化合物组合构成100%。它还含有0-1000重量ppm的硫,优选0-500重量ppm的硫。
[0150]
优选地,根据本发明的选择性加氢方法处理的多不饱和烃原料是蒸汽裂化c2馏分、或蒸汽裂化c2-c3馏分、或蒸汽裂化汽油。
[0151]
根据本发明的选择性加氢方法旨在除去待加氢的所述原料中存在的所述多不饱和烃,而不将单不饱和烃加氢。例如,当所述原料是c2馏分时,选择性加氢方法旨在将乙炔选择性加氢。当所述原料是c3馏分时,选择性加氢方法旨在将丙二烯和甲基乙炔选择性加氢。在c4馏分的情况下,目标是除去丁二烯、乙烯基乙炔(vac)和丁炔;在c5馏分的情况下,目标是除去戊二烯。当所述原料是蒸汽裂化汽油时,选择性加氢方法旨在将存在于所述待处理的原料中的所述多不饱和烃选择性加氢,从而将二烯烃化合物部分加氢以产生单烯烃,和从而将苯乙烯类化合物和茚类化合物部分加氢以产生相应的芳族化合物,同时避免芳环的加氢。
[0152]
选择性加氢方法的技术实施通过例如将多不饱和烃原料和氢气上流式或下流式地注入至少一个固定床反应器中来实施。所述反应器可以是等温型或绝热型。绝热反应器是优选的。多不饱和烃原料可有利地通过将由发生选择性加氢反应的所述反应器获得的流出物在位于反应器的入口和出口之间的反应器的各种点处的一个或多个再注入来稀释,以限制反应器中的温度梯度。根据本发明的选择性加氢方法的技术实施还可以有利地通过在反应性蒸馏塔或反应器-交换器中或在浆料型反应器中装入至少所述负载型催化剂来实施。氢气流可以与待加氢的原料同时引入和/或在反应器的一个或多个不同点处引入。
[0153]
蒸汽裂化c2馏分、c2-c3馏分、c3馏分、c4馏分、c5馏分和c5 馏分的选择性加氢可以在气相或液相中实施,对于c3馏分、c4馏分、c5馏分和c5 馏分而言,优选在液相中实施,对于c2馏分和c2-c3馏分而言,优选在气相中实施。液相反应可以降低能量成本并延长催化剂的循环时间。
[0154]
通常,包含每分子含有至少2个碳原子的多不饱和化合物且终沸点低于或等于300℃的烃原料的选择性加氢在0-300℃的温度、0.1-10mpa的压力下,对于在液相中实施的方法,在0.1-10的氢气/(待加氢的多不饱和化合物)摩尔比以及0.1-200h-1
的时空速(定义为原料的体积流量与催化剂的体积之比)下实施,或者对于在气相中实施的方法,在0.5-1000的氢气/(待加氢的多不饱和化合物)摩尔比以及100-40000h-1
的时空速下实施。
[0155]
在根据本发明的一个实施方案中,当实施其中原料是包含多不饱和化合物的蒸汽裂化汽油的选择性加氢方法时,(氢气)/(待加氢的多不饱和化合物)摩尔比通常为0.5-10,优选0.7-5.0,更优选1.0-2.0,温度为0-200℃,优选20-200℃,更优选30-180℃,时空速
(hsv)通常为0.5-100h-1
,优选1-50h-1
,压力通常为0.3-8.0mpa,优选1.0-7.0mpa,更优选1.5-4.0mpa。
[0156]
更优选地,实施其中原料是包含多不饱和化合物的蒸汽裂化汽油的选择性加氢方法,氢气/(待加氢的多不饱和化合物)摩尔比为0.7-5.0,温度为20-200℃,时空速(hsv)通常为1-50h-1
,压力为1.0-7.0mpa。
[0157]
还更优选地,实施其中原料是包含多不饱和化合物的蒸汽裂化汽油的选择性加氢方法,氢气/(待加氢的多不饱和化合物)摩尔比为1.0-2.0,温度为30-180℃,时空速(hsv)通常为1-50h-1
,压力为1.5-4.0mpa。
[0158]
调节氢气流量以使其具有足以在理论上将所有多不饱和化合物加氢并在反应器出口处保持氢气过量的量。
[0159]
在根据本发明的另一个实施方案中,当实施其中原料是包含多不饱和化合物的蒸汽裂化c2馏分和/或蒸汽裂化c2-c3馏分的选择性加氢方法时,(氢气)/(待加氢的多不饱和化合物)摩尔比通常为0.5-1000,优选0.7-800,温度为0-300℃,优选15-280℃,时空速(hsv)通常为100-40000h-1
,优选500-30000h-1
,压力通常为0.1-6.0mpa,优选0.2-5.0mpa。
[0160]
芳族化合物加氢方法本发明的另一个主题是对终沸点低于或等于650℃、通常为20℃-650℃、优选为20℃-450℃的烃原料中含有的至少一种芳族或多芳族化合物进行加氢的方法。含有至少一种芳族或多芳族化合物的所述烃原料可以选自以下石油或石化馏分:催化重整产生的重整产物、煤油、轻质瓦斯油、重质瓦斯油、裂化馏出物,例如fcc再循环油、焦化单元瓦斯油或加氢裂化馏出物。
[0161]
在根据本发明的加氢方法中处理的烃原料中含有的芳族或多芳族化合物的含量通常为0.1-80重量%,优选1-50重量%,特别优选2-35重量%,所述百分数基于烃原料的总重量。存在于所述烃原料中的芳族化合物是,例如,苯或烷基芳族化合物,例如甲苯、乙苯、邻二甲苯、间二甲苯或对二甲苯,或者具有多个芳环的芳族化合物(多芳族化合物),例如萘。
[0162]
原料中的硫或氯的含量通常低于5000重量ppm,优选低于100重量ppm,特别优选低于10重量ppm的硫或氯。
[0163]
芳族或多芳族化合物的加氢方法的技术实施通过例如将烃原料和氢气上流式或下流式地注入至少一个固定床反应器中来实施。所述反应器可以是等温型或绝热型。绝热反应器是优选的。烃原料可有利地通过将由发生芳族化合物的加氢反应的所述反应器获得的流出物在位于反应器的入口和出口之间的反应器的各种点处的一个或多个再注入来稀释,以限制反应器中的温度梯度。根据本发明的芳族化合物的加氢方法的技术实施还可以有利地通过在反应性蒸馏塔或反应器-交换器中或在浆料型反应器中装入至少所述负载型催化剂来实施。氢气流可以与待加氢的原料同时引入和/或在反应器的一个或多个不同点处引入。
[0164]
芳族或多芳族化合物的加氢可以在气相或液相中进行,优选在液相中进行。通常,含有芳族或多芳族化合物且终沸点低于或等于650℃、通常为20℃-650℃、优选为20℃-450℃的烃原料的芳族或多芳族化合物的加氢在30℃-350℃、优选50℃-325℃的温度、在0.1-20mpa、优选0.5-10mpa的压力、0.1-10的氢气/(待加氢的芳族化合物)摩尔比、在0.05-50h-1
、优选0.1-10h-1
的时空速下进行。
[0165]
调节氢气流量以使其具有足以在理论上将所有芳族化合物加氢并在反应器出口处保持氢气过量的量。
[0166]
芳族或多芳族化合物的转化率通常大于20摩尔%,优选大于40摩尔%,更优选大于80摩尔%,特别优选大于90摩尔%的烃原料中所含的芳族或多芳族化合物。通过用烃原料中的芳族或多芳族化合物的总摩尔数与产物中的芳族或多芳族化合物的总摩尔数之间的差值除以烃原料中的芳族或多芳族化合物的总摩尔数计算转化率。
[0167]
根据本发明方法的一种特定的替代形式,进行将烃原料(例如由催化重整单元产生的重整产物)中的苯加氢的方法。所述烃原料中的苯含量通常为0.1-40重量%,优选为0.5-35重量%,特别优选为2-30重量%,所述重量百分数基于烃原料的总重量。
[0168]
原料中的硫或氯的含量通常各自小于10重量ppm,优选小于2重量ppm的硫或氯。
[0169]
烃原料中所含的苯的加氢可以在气相或液相中进行,优选在液相中进行。当它在液相中进行时,可以存在溶剂,例如环己烷、庚烷或辛烷。通常,苯的加氢在30℃-250℃、优选50℃-200℃、更优选80℃-180℃的温度、在0.1-10mpa、优选0.5-4mpa的压力、0.1-10的氢气/(苯)摩尔比、在0.05-50h-1
、优选0.5-10h-1
的时空速下进行。
[0170]
苯的转化率通常大于50mol%,优选大于80mol%,更优选大于90mol%,特别优选大于98mol%。
[0171]
现在将通过以下实施例来举例说明本发明,所述实施例绝不是限制性的。
实施例
[0172]
实施例1:氧化铝al-1的制备通过铝酸钠和硫酸铝的混合物合成氧化铝凝胶。沉淀反应在60℃的温度、9的ph下进行60分钟,搅拌速度为200rpm。
[0173]
将由此获得的凝胶在z型臂混合机中捏合,以形成糊料。通过使糊料通过配备有直径为1.6mm的三叶形孔的模头来进行挤出。将由此获得的挤出物在干燥空气流下在150℃下干燥12小时,然后在干燥空气流下在450℃下煅烧5小时。
[0174]
在含有相对于氧化铝重量为6.5重量%的乙酸的水溶液的存在下、在高压釜中、在650℃下对挤出物进行水热处理3小时,然后在管状反应器中、在1000℃下在干燥空气中煅烧2小时。获得氧化铝al-1。
[0175]
氧化铝al-1的比表面积为80m
²
/g,孔体积(由hg测孔仪测定)为0.85ml/g,中孔直径为35nm。
[0176]
相对于氧化铝的总重量,钠含量为0.0350重量%,相对于氧化铝的总重量,硫含量为0.15重量%。
[0177]
实施例1a:氧化铝al-2的制备通过铝酸钠和硫酸铝的混合物合成氧化铝凝胶。沉淀反应在60℃的温度、9的ph下进行60分钟,搅拌速度为200rpm。将由此获得的凝胶在z型臂混合机中捏合,以形成糊料。通过使糊料通过配备有直径为1.6mm的三叶形孔的模头来进行挤出。将由此获得的挤出物在干燥空气流下在150℃下干燥12小时,然后在干燥空气流下在450℃下煅烧5小时。获得氧化铝al-2。
[0178]
氧化铝al-2的比表面积为255m
²
/g,孔体积(由hg测孔仪测定)为0.7ml/g,中值中
孔直径为12nm。
[0179]
相对于氧化铝的总重量,钠含量为0.0350重量%,相对于氧化铝的总重量,硫含量为0.15重量%。
[0180]
实施例2:ni前体的水溶液的制备用于制备催化剂d的ni前体的水溶液(溶液s1)通过将43.5g的硝酸镍(nino3,供应商strem chemicals
®
)溶解在体积为13ml的蒸馏水中来制备。
[0181]
获得溶液s1,其镍浓度为每升溶液350g的ni。
[0182]
实施例3:具有根据步骤d2)的有机化合物的ni前体的水溶液的制备用于制备催化剂a、催化剂b、催化剂c、催化剂e和催化剂f的ni前体的水溶液(溶液s2)通过将43.5g的硝酸镍(nino3,供应商strem chemicals
®
)和丙二酸(cas 141-82-2;供应商fluka
®
)溶解在体积为13ml的蒸馏水中来制备。丙二酸/镍的摩尔比为0.5。
[0183]
实施例3a:具有根据步骤d2)的有机化合物的ni前体的第二水溶液的制备用于制备催化剂a、催化剂b、催化剂c、催化剂e和催化剂f的ni前体的水溶液(溶液s2b)通过将14.5g的硝酸镍(nino3,供应商strem chemicals
®
)和丙二酸(cas 141-82-2;供应商fluka
®
)溶解在体积为13ml的蒸馏水中来制备。丙二酸/镍的摩尔比为0.5。
[0184]
获得溶液s2b,其镍浓度为每升溶液116g的ni。
[0185]
实施例4:根据本发明的催化剂a的制备(根据本发明)将实施例3中制备的溶液s2通过逐滴添加的方式干法浸渍在10g的根据实施例1获得的氧化铝al-1上。
[0186]
随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。在该实施例和下文所有的实施例中使用的干燥空气包含小于5克的水/千克干燥空气。
[0187]
用含甲酸的水溶液干法浸渍由此获得的催化剂前体,hcooh/ni的摩尔比等于1mol/mol。
[0188]
在含甲酸的水溶液的浸渍结束后,在每千克干燥空气中含有50克水且流量为1升/小时/克催化剂的空气流下、在150℃下对催化剂前体进行热处理2小时,然后在干燥空气流下、在120℃下进行1小时。
[0189]
获得含有相对于催化剂总重量为15重量%的镍元素的催化剂a。由此获得的催化剂a的特性报道在下文的表1中。
[0190]
实施例5:根据本发明的催化剂b的制备(根据本发明)将实施例3a中制备的溶液s2b通过逐滴添加的方式干法浸渍在10g的根据实施例1获得的氧化铝al-1上。该步骤中的目标镍含量是最终催化剂上的5重量%的镍。随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。
[0191]
用含甲酸的水溶液干法浸渍由此获得的催化剂前体,hcooh/ni的摩尔比等于1mol/mol。
[0192]
在含甲酸的水溶液的浸渍结束后,在每千克干燥空气中含有50克水且流量为1升/小时/克催化剂的空气流下、在150℃下对催化剂前体进行热处理2小时,然后在干燥空气流下、在120℃下进行1小时。
[0193]
获得含有相对于催化剂总重量为5重量%的镍元素的催化剂b。由此获得的催化剂b的特性报道在下文的表1中。
[0194]
实施例6:根据本发明的催化剂c的制备(根据本发明)将实施例3中制备的溶液s2通过逐滴添加的方式干法浸渍在10g的根据实施例1获得的氧化铝al-1上。
[0195]
随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。
[0196]
用含乙醇酸的水溶液干法浸渍由此获得的催化剂前体。c2h4o3/ni的比率等于2mol/mol。
[0197]
在含乙醇酸的水溶液的浸渍结束后,在每千克干燥空气中含有50克水且流量为1升/小时/克催化剂的空气流下、在150℃下对催化剂前体进行热处理2小时,然后在干燥空气流下、在120℃下进行1小时。
[0198]
获得含有相对于催化剂总重量为15重量%的镍元素的催化剂c。由此获得的催化剂c的特性报道在下文的表1中。
[0199]
实施例7:催化剂d的制备(不根据本发明)将实施例2中制备的溶液s1通过逐滴添加的方式干法浸渍在10g的根据实施例1获得的氧化铝al-1上。
[0200]
随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。
[0201]
用含甲酸的水溶液干法浸渍由此获得的催化剂前体。hcooh/ni的比率等于1mol/mol。
[0202]
在含甲酸的水溶液的浸渍结束后,在每千克干燥空气中含有50克水且流量为1升/小时/克催化剂的空气流下、在150℃下对催化剂前体进行热处理2小时,然后在干燥空气流下、在120℃下进行1小时。
[0203]
获得含有相对于催化剂总重量为15重量%的镍元素的催化剂d。由此获得的催化剂d的特性报道在下文的表1中。
[0204]
实施例8:催化剂e的制备(不根据本发明)将实施例3中制备的溶液s2通过逐滴添加的方式干法浸渍在10g的根据实施例1a获得的氧化铝al-2上。
[0205]
随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。
[0206]
用含甲酸的水溶液干法浸渍由此获得的催化剂前体,hcooh/ni的比率等于1mol/mol。
[0207]
在含甲酸的水溶液的浸渍结束后,在每千克干燥空气中含有50克水且流量为1升/小时/克催化剂的空气流下、在150℃下对催化剂前体进行热处理2小时,然后在干燥空气流下、在120℃下进行1小时。
[0208]
获得含有相对于催化剂总重量为15重量%的镍元素的催化剂e。由此获得的催化剂e的特性报道在下文的表1中。
[0209]
实施例9:催化剂f的制备(不根据本发明)
将实施例3中制备的溶液s2通过逐滴添加的方式干法浸渍在10g的根据实施例1获得的氧化铝al-1上。
[0210]
随后将由此获得的固体在120℃下在烘箱中干燥12小时,然后在1升/小时/克催化剂的干燥空气流下、在450℃下煅烧2小时。
[0211]
获得含有相对于催化剂总重量为15重量%的镍元素的催化剂f。由此获得的催化剂f的特性报道在下文的表1中。表1:催化剂a至f的特性。
[0212]
实施例10:催化测试:含有苯乙烯和异戊二烯的混合物的选择性加氢性能(a
hyd1
)针对含有苯乙烯和异戊二烯的混合物的选择性加氢反应,对上述实施例中描述的催化剂a至催化剂f进行了测试。
[0213]
待选择性加氢的原料的组成如下:8重量%的苯乙烯(由sigma aldrich
®
提供,纯度为99%)、8重量%的异戊二烯(由sigma aldrich
®
提供,纯度为99%)和84重量%的正庚烷(溶剂)(由vwr
®
提供,纯度》99% chromanorm hplc)。该原料还含有极低含量的含硫化合物:以戊硫醇(由fluka
®
提供,纯度》 97%)形式引入的10重量ppm的硫和以噻吩(由merck
®
提供,纯度99%)形式引入的100重量ppm的硫。该组成相当于反应混合物的初始组成。这种模型分子的混合物代表裂解汽油。
[0214]
选择性加氢反应在500ml的不锈钢高压釜中进行,该高压釜配备有磁力驱动的机械搅拌器,并且能够在100巴(10mpa)的最大压力和5℃-200℃的温度下操作。
[0215]
在将催化剂引入高压釜之前,将3ml量的催化剂在1升/小时/克催化剂的氢气流下在400℃下非原位还原16小时(温升梯度为1℃/分钟),然后将其免受空气影响地转移至高压釜。在加入214ml的正庚烷(由vwr
®
提供,纯度》99% chromanorm hplc)后,关闭高压釜,进行吹扫,然后在35巴(3.5mpa)的氢气下加压并使高压釜达到测试温度,等于30℃。在时间t = 0时,将约30g的含有苯乙烯、异戊二烯、正庚烷、戊硫醇和噻吩的混合物引入高压釜。然后,反应混合物具有上述组成并以1600rpm开始搅拌。使用位于反应器上游的储气罐将高压釜内的压力保持恒定在35巴(3.5mpa)。
[0216]
通过以规律的时间间隔从反应介质中取样来监测反应的进展:苯乙烯被加氢以产
生乙苯,而芳环没有被加氢,异戊二烯被加氢以产生甲基丁烯。如果反应延长超过了必要的时间,则甲基丁烯进而被加氢以产生异戊烷。也通过位于反应器上游的储气罐中的压力的降低随时间监测氢气的消耗。催化活性以每分钟和每克ni消耗的h2的摩尔数表示。
[0217]
下表2报道了测定的催化剂a至催化剂f的催化活性。将它们与对催化剂e测得的催化活性(a
hyd1
)相关联。
[0218]
实施例11:催化测试:甲苯加氢的性能(a
hyd2
)针对甲苯的加氢反应,还对上述实施例中描述的催化剂a至催化剂f进行了测试。
[0219]
选择性加氢反应在与实施例10中描述的高压釜相同的高压釜中进行。
[0220]
在将催化剂引入高压釜之前,将2ml量的催化剂在1升/小时/克催化剂的氢气流下在400℃下非原位还原16小时(温升梯度为1℃/分钟),然后将其免受空气影响地转移至高压釜。在加入216ml的正庚烷(由vwr
®
提供,纯度》99% chromanorm hplc)后,关闭高压釜,进行吹扫,然后在35巴(3.5mpa)的氢气下加压,并使高压釜达到测试温度,等于80℃。在时间t = 0时,将约26g的甲苯(由sds
®
提供,纯度》99.8%)引入高压釜(反应混合物的初始组成为6wt%甲苯/94wt%正庚烷),并以1600rpm开始搅拌。使用位于反应器上游的储气罐将高压釜内的压力保持恒定在35巴(3.5mpa)。
[0221]
通过以规律的时间间隔从反应介质中取样来监测反应的进展:甲苯被完全加氢以产生甲基环己烷。也通过位于反应器上游的储气罐中的压力的降低随时间监测氢气的消耗。催化活性以每分钟和每克ni消耗的h2的摩尔数表示。
[0222]
下表2报道了测定的催化剂a至催化剂f的催化活性。将它们与对催化剂e测得的催化活性(a
hyd2
)相关联。催化剂ni
°
含量(%)a
hyd1
(%)a
hyd2
(%)a(根据本发明)15180175b(根据本发明)5120117c(根据本发明)15195190d(不根据本发明)159585e(不根据本发明)15100100f(不根据本发明)159189表2:催化剂a至催化剂f在含有苯乙烯和异戊二烯的混合物的选择性加氢中的性能(a
hyd1
)和在甲苯的加氢中的性能(a
hyd2
)的对比。
[0223]
这清楚地表明,与不根据本发明的催化剂d至f相比,根据本发明的催化剂a、b和c的性能改善。催化剂b的ni含量是催化剂d、e和f的三分之一,并且由于其小的 ni 颗粒及其在壳体中的分布,与具有遍布催化剂粒子均匀分布的15%ni的催化剂相比,催化剂 b 的活性改善。由于使用不含添加剂的溶液s1(这导致比根据本发明更大的颗粒尺寸,尽管分布在壳体中),催化剂d具有较低的活性。与本发明相比,催化剂e和f的制备不同。对于催化剂e,使用的载体是不根据本发明的氧化铝al-2,对于催化剂f,没有进行步骤e)和f)。在这两种情况下,镍遍布催化剂粒子均匀分布。 因此催化剂e和f在a
hyd1
和a
hyd2
方面具有比催化剂a低得多的活性。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。