1.本发明涉及医药技术领域,具体涉及原人参二醇类化合物在治疗疼痛和成瘾物质躯体、精神依赖和成瘾的用途。
背景技术:
2.疼痛,是一种与伤害及痛苦相关联的令人厌恶的复合感受。在正常生理条件下,疼痛能够在躯体收到威胁时提供报警信号,是一种不可或缺的生命保护功能。但在病理条件下,疼痛是大多数疾病具有的共同症状,与自主神经活动、运动反射、心理和情绪反应等交织在一起,给患者带来痛苦。
3.根据起因、性质、部位和时程的不同,疼痛分为急性疼痛和慢性疼痛。急性疼痛是指生理状态下,伤害性刺激直接激活相应部位的伤害性感受器而引起的疼痛。急性疼痛持续时间不长(《1月),在损伤修复后,疼痛则自行消失。急性疼痛包括术后疼痛、创伤后疼痛、急性头疼和面部疼痛、急性关节炎疼痛等。
4.慢性疼痛则是在病灶修复后,疼痛依然持续存在,可长达数月(》1月)甚至终生,或可经常复发。慢性疼痛包括下背部疼痛、癌症疼痛、抗肿瘤药物和阿片类药物引起的疼痛、糖尿病性疼痛、神经病理性疼痛包括带状疱疹后遗神经痛、三叉神经痛和坐骨神经痛、炎性疼痛、幻肢痛、关节炎疼痛、纤维肌痛、肌肉骨骼疼痛、慢性区域性疼痛综合征、创伤后神经痛和周围神经病等。
5.根据国际疼痛研究学会(international association for the study of pain,iasp)数据,全世界慢性疼痛的患病率为10%(scholz et al.,pain,160:53-59,2019)。仅在西欧,据报道就有8.0%的人口患有慢性疼痛。在中国,约有20%的糖尿病患者被诊断为糖尿病周围神经病,三分之一的带状疱疹病毒患者发展为带状疱疹后遗神经痛,超过一百万的人患有癌性疼痛。疼痛给患者带来痛苦,严重影响了家庭生活或工作,也给公共卫生系统带来巨大负担。
6.常用治疗疼痛药物有:1)非甾体抗炎镇痛药物包括氟比洛芬酯、布洛芬、双氯芬酸钠、美洛昔康、萘普生、塞来昔布和普瑞昔布;
7.2)抗癫痫药物包括卡马西平、苯妥英钠和加巴喷丁类药物如加巴喷丁、普瑞巴林和米罗巴林;
8.3)单胺类神经递质重摄取抑制剂抗抑郁药包括阿米替林和度洛西丁;
9.4)局部麻醉药包括利多卡因、罗哌卡因、丙胺卡因;
10.5)阿片类镇痛药包括可待因、双氢可待因、吗啡、芬太尼、舒芬太尼、瑞芬太尼、哌替啶、羟考酮;
11.6)去甲肾上腺素α2受体激动剂如可乐定、右美托咪定;
12.7)mor-nri双靶点镇痛药如地佐辛、他喷他多、喷他佐辛和曲马多;
13.8)中草药包括独一味、乌头/附子及其有效成分如草乌甲素和高乌甲素,以及延胡
索及其有效成分如罗通定。
14.但这些药物治疗疼痛或疗效有限或产生较多较重不良反应。如加巴喷丁和瑞普巴林不是特异性镇痛药,其治疗神经病理性疼痛的有效(降低疼痛阈值30%)人群低于50%。非甾体类镇痛药对头痛、牙痛、肌肉和关节痛等有一定疗效,而对外伤剧痛和内脏平滑肌绞痛几乎无效。局部麻醉药仅适用于外周神经病理性疼痛。
15.再如阿片类药物有多种不良反应如嗜睡、呼吸抑制、便秘等,长期服用会产生镇痛耐受、痛觉过敏、躯体依赖性和成瘾及滥用。加巴喷丁和瑞普巴林也有嗜睡等严重不良反应。反复使用阿片类药物包括吗啡和芬太尼,机体会产生镇痛耐受,必须增加剂量才能获得相同的镇痛效果。长期或反复使用阿片类药物也可导致成瘾,包括躯体依赖(身体依赖、生理依赖)和精神依赖(心理依赖)两个方面。
16.躯体依赖性是为避免戒断症状而反复用药,且由于耐受性剂量逐渐加大,在成瘾过程中表现为厌恶效应,起到负强化效应。精神依赖性是指依赖者的心理觅药的渴求和重复用药达到的欣快感,表现为奖赏作用,起到正强化效应,促使患者屡次复吸。目前解决阿片类药物成瘾方法和途径仍有较大局限,美沙酮、丁丙诺啡、可乐定、洛非西定等仅在一定程度上改善戒断症状,疗效十分有限,尤其对精神依赖几无疗效。
17.因此,疼痛治疗目前仍然是临床一大难题,急需研发可长期使用且自身无镇痛耐受和成瘾性,能有效治疗疼痛和阿片类药物诱导的躯体和精神依赖的新型镇痛药物。
技术实现要素:
18.本发明的目的是提供一种可长期使用且自身无镇痛耐受和成瘾性,能有效治疗疼痛和阿片类药物诱导的躯体和精神依赖的新型镇痛药物。
19.具体地,本发明提供了原人参二醇类化合物(如20(s)-原人参二醇)在制备治疗疼痛和阿片类药物诱导的躯体和精神依赖的新型镇痛药物中的应用。本发明实验表明,20(s)-原人参二醇还可以和其他活性成分联用,达到协同镇痛的作用。并且,20(s)-原人参二醇通过激动脊髓小胶质细胞糖皮质激素受体(细胞膜糖皮质激素受体)表达和释放强啡肽a,从而产生镇痛作用。
20.在本发明的第一方面,提供了一种活性成分或含所述活性成分的制剂的用途,所述活性成分选自下组:原人参二醇或其药学上可接受的盐或酯、原人参三醇或其药学上可接受的盐或酯;
21.并且,所述活性成分或含所述活性成分的制剂用于制备:
22.(a)治疗和/或缓解疼痛的药物;和/或
23.(b)治疗和/或缓解成瘾物质诱导的躯体和/或精神依赖和/或成瘾的药物。
24.在另一优选例中,所述的原人参二醇包括20(s)-原人参二醇、20(r)-原人参二醇、或其组合。
25.在另一优选例中,所述的原人参三醇包括20(s)-原人参三醇、20(r)-原人参三醇、或其组合。
26.在另一优选例中,所述成瘾物质选自:阿片类药物、海洛因、或其组合。
27.在另一优选例中,所述成瘾物质还包括选自下组的一种或多种:冰毒、酒精、香烟(尼古丁)、可卡因、大麻、或其组合。
28.在另一优选例中,所述疼痛选自:神经病理性疼痛、炎症性疼痛、关节炎疼痛、糖尿病性疼痛、下背部疼痛、脊髓损伤性疼痛、内脏疼痛、纤维肌痛、慢性局部疼痛综合征、肌肉骨骼疼痛、癌症疼痛、抗肿瘤药物和阿片类药物引起的疼痛、手术后疼痛、创伤后疼痛、创伤后神经痛和周围神经病、幻肢痛、或其组合。
29.在另一优选例中,所述神经病理性疼痛包括(但不限于)带状疱疹后遗神经痛、三叉神经痛和坐骨神经痛。
30.在另一优选例中,所述制剂还包括第二活性成分;其中第二活性成分选自下组:
31.(z1)选自下组的阿片类镇痛药:可待因、双氢可待因、吗啡、芬太尼、舒芬太尼、瑞芬太尼、哌替啶、羟考酮或其组合;
32.(z2)选自下组的抗癫痫药物:卡马西平、苯妥英钠、加巴喷丁类(gabapentinoids)化合物、或其组合;
33.(z3)选自下组的非甾体抗炎镇痛药物:氟比洛芬酯、布洛芬、双氯芬酸钠、美洛昔康、萘普生、塞来昔布、普瑞昔布或其组合;
34.(z4)选自下组的单胺类神经递质重摄取抑制剂抗抑郁药:阿米替林、度洛西丁、或其组合;
35.(z5)选自下组的局部麻醉药:利多卡因、罗哌卡因、丙胺卡因、或其组合;
36.(z6)选自下组的去甲肾上腺素α2受体激动剂:可乐定、右美托咪定、或其组合;
37.(z7)选自下组的mor-nri双靶点镇痛药:地佐辛、他喷他多、喷他佐辛、曲马多、或其组合;
38.(z8)选自下组的抗偏头痛药:cgrp抗体及其受体拮抗剂;
39.(z9)选自下组的中草药:独一味提取物及其有效成分、乌头/附子及其有效成分、以及延胡索及其有效成分、或其组合;
40.(z10)上述z1~z9的任意组合。
41.在另一优选例中,所述加巴喷丁类化合物包括巴喷丁、普瑞巴林和米罗巴林(mirogabalin)。
42.在另一优选例中,所述独一味提取物及其有效成分包括山栀子苷甲酯、8-o-乙酰山栀子苷甲酯。
43.在另一优选例中,所述乌头/附子及其有效成分包括:草乌甲素、高乌甲素和雪上一支蒿甲素。
44.在另一优选例中,所述延胡索及其有效成分包括四氢巴马汀、千金藤啶碱、紫堇达明和脱氢紫堇鳞茎碱。
45.在另一优选例中,所述制剂为口服制剂、或注射剂。
46.在另一优选例中,所述制剂包括:粉剂、颗粒剂、胶囊剂、针剂、酊剂、口服液、片剂、含片、或滴丸。
47.在另一优选例中,所述活性成分或含所述活性成分的制剂不具有:(1)镇痛耐受性;(2)躯体依赖性;(3)精神依赖性(成瘾性)。
48.在本发明的第二方面,提供了一种药物组合物,所述药物组合物包含:
49.(i)第一活性成分,所述第一活性成分选自下组:原人参二醇、原人参三醇;
50.(ii)第二活性成分,所述第二活性成分选自:
51.(z1)选自下组的阿片类镇痛药:可待因、双氢可待因、吗啡、芬太尼、舒芬太尼、瑞芬太尼、哌替啶、羟考酮或其组合;
52.(z2)选自下组的抗癫痫药物:卡马西平、苯妥英钠、加巴喷丁类化合物、或其组合;
53.(z3)选自下组的非甾体抗炎镇痛药物:氟比洛芬酯、布洛芬、双氯芬酸钠、美洛昔康、萘普生、塞来昔布、普瑞昔布或其组合;
54.(z4)选自下组的单胺类神经递质重摄取抑制剂抗抑郁药:阿米替林、度洛西丁、或其组合;
55.(z5)选自下组的局部麻醉药:利多卡因、罗哌卡因、丙胺卡因、或其组合;
56.(z6)选自下组的去甲肾上腺素α2受体激动剂:可乐定、右美托咪定、或其组合;
57.(z7)选自下组的mor-nri双靶点镇痛药:地佐辛、他喷他多、喷他佐辛、曲马多、或其组合;
58.(z8)选自下组的抗偏头痛药:cgrp抗体及其受体拮抗剂;
59.(z9)选自下组的中草药:独一味提取物及其有效成分、乌头/附子及其有效成分、以及延胡索及其有效成分、或其组合;
60.(z10)上述z1~z9的任意组合;
61.(iii)药学上可接受的载体和/或赋形剂。
62.在另一优选例中,所述药物组合物给药方式是口服的或非口服的。
63.在另一优选例中,所述非口服的给药方式选自下组:鼻饲、肛门栓塞、皮下注射、肌肉注射、静脉注射、蛛网膜下腔注射、硬膜外注射,侧脑室注射、皮肤外敷(贴剂)、或其组合。
64.在本发明的第三方面,提供了一种第二方面所述的药物组合物的用途,所述药物组合物用于制备:
65.(a)治疗和/或缓解疼痛的药物;
66.(b)治疗和/或缓解成瘾物质诱导的躯体和/或精神依赖的药物。
67.在本发明的第四方面,提供了一种体外的治疗和/或缓解疼痛的方法,包括步骤:
68.(1)在含有有效量的第一活性成分或含所述活性成分的制剂或如第二方面所述的药物组合物的体系中培养细胞,其中所述第一活性成分选自下组:原人参二醇或其药学上可接受的盐或酯、原人参三醇或其药学上可接受的盐或酯;
69.(2)激动所述细胞糖皮质激素受体表达强啡肽a。
70.在另一优选例中,所述细胞为中枢神经系统的免疫细胞,优选地脊髓免疫细胞。
71.在另一优选例中,所述细胞选自:小胶质细胞、巨噬细胞、单核细胞、或其组合。
72.在另一优选例中,所述细胞为脊髓小胶质细胞。
73.在另一优选例中,所述糖皮质激素为糖皮质激素受体激动剂。
74.在另一优选例中,所述糖皮质激素为细胞膜糖皮质激素受体激动剂。
75.在另一优选例中,所述方法为非诊断性非治疗性的。
76.在本发明的第五方面,提供了一种治疗和/或缓解疼痛的方法,包括步骤:
77.给有需要的对象施用医学有效量的第一活性成分或含所述活性成分的制剂或第二方面所述的药物组合物,其中所述第一活性成分选自下组:原人参二醇或其药学上可接受的盐或酯、原人参三醇或其药学上可接受的盐或酯;从而治疗和/或缓解疼痛。
78.在本发明的第六方面,提供了一种诱导产生强啡肽a表达和释放的方法,包括:给
需要的对象施用活性成分,所述活性成分选自下组:原人参二醇或其药学上可接受的盐或酯、原人参三醇或其药学上可接受的盐或酯,从而诱导所述对象产生强啡肽a。
79.在另一优选例中,所述的方法刺激脊髓增加强啡肽a的表达和释放。
80.在另一优选例中,所述方法用于激动所述对象细胞的糖皮质激素受体。
81.在另一优选例中,所述糖皮质激素受体为细胞膜糖皮质激素受体。
82.在另一优选例中,所述对象为哺乳动物。
83.在另一优选例中,所述对象包括但不限于老鼠、人。
84.在另一优选例中,所述对象为疼痛患者。
85.在另一优选例中,所述细胞为中枢神经系统的免疫细胞,优选地脊髓免疫细胞。
86.在另一优选例中,所述细胞选自:小胶质细胞、巨噬细胞、单核细胞、或其组合。
87.在另一优选例中,所述细胞为脊髓小胶质细胞。
88.在另一优选例中,所述方法为非诊断性和非治疗性的。
89.应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
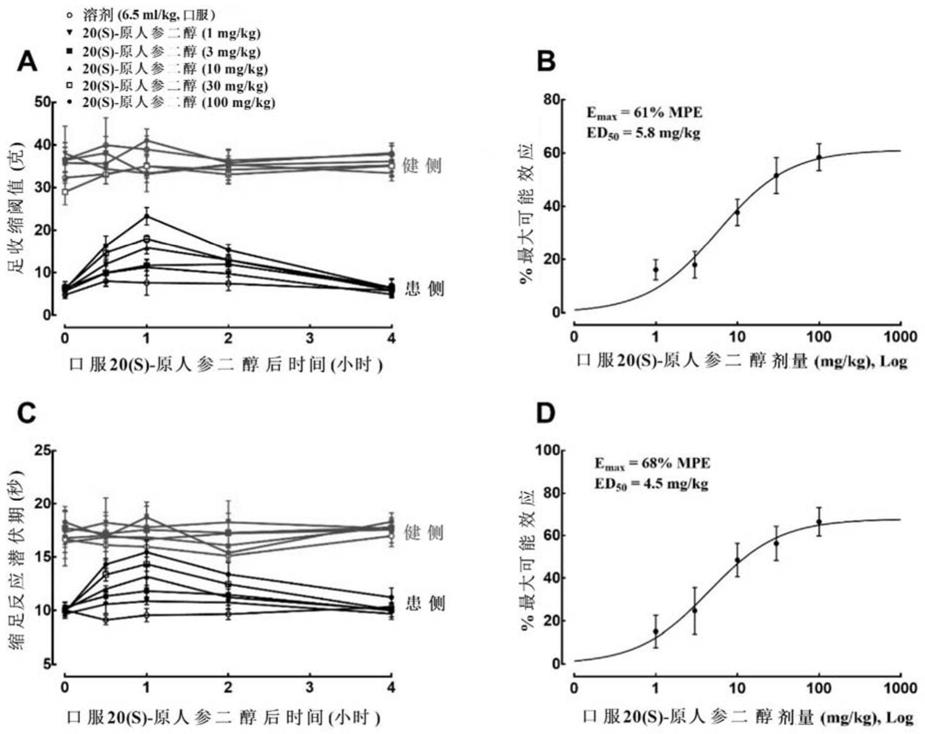
90.图1表明口服20(s)-原人参二醇呈剂量依赖式地抑制神经病理性疼痛的机械痛敏和热痛敏。
91.图2显示口服20(s)-原人参二醇在骨癌疼痛、完全弗氏佐剂(cfa)炎症性疼痛和福尔马林疼痛模型中镇痛作用。
92.图3显示口服20(s)-原人参二醇联合加巴喷丁或吗啡在神经病理性疼痛模型中协同镇痛作用。
93.图4显示口服20(s)-原人参二醇特异性地刺激大鼠脊髓表达强啡肽a基因和蛋白质。
94.图5免疫荧光双染色显示口服20(s)-原人参二醇特异性地刺激大鼠脊髓小胶质细胞表达强啡肽a。
95.图6显示离体给予20(s)-原人参二醇特异性地刺激原代脊髓小胶质细胞表达强啡肽a基因和蛋白。
96.图7显示小胶质细胞活化抑制剂米诺环素阻断20(s)-原人参二醇抗神经病理性疼痛。
97.图8显示强啡肽a抗血清和特异性κ-阿片受体拮抗剂阻断20(s)-原人参二醇抗神经病理性疼痛。
98.图9显示口服20(s)-原人参二醇不产生自身镇痛耐受性,但抑制吗啡镇痛耐受作用。
99.图10显示口服20(s)-原人参二醇不产生躯体依赖性,但抑制吗啡躯体依赖作用。
100.图11显示口服20(s)-原人参二醇不产生精神依赖性,但抑制吗啡精神依赖作用。
101.图12显示蛛网膜下腔鞘内预先给予糖皮质激素受体拮抗剂抑制口服20(s)-原人参二醇的镇痛作用。
102.图13显示蛛网膜下腔鞘内预先给予糖皮质激素受体拮抗剂抑制口服20(s)-原人参二醇在脊髓所产生的强啡肽a表达作用。
103.图14显示糖皮质激素受体拮抗剂抑制20(s)-原人参二醇在原代脊髓小胶质细胞所产生的强啡肽a表达作用。
具体实施方式
104.本发明人经过广泛而深入地研究,首次意外发现,原人参二醇类化合物(尤其是20(s)-原人参二醇)可显著地抑制疼痛,并具有明显的抗成瘾作用,并且不产生镇痛耐受性、躯体依赖性、精神依赖性等不良反应。因此可用于治疗疼痛和抗成瘾(如成瘾物质诱导的躯体和精神依赖)。在此基础上完成本发明。
105.具体地,实施例表明,活性成分20(s)-原人参二醇的镇痛作用不产生自身镇痛耐受性,并且能在镇痛同时帮助抑制成瘾物质诱导的躯体依赖、精神依赖作用。原人参二醇类化合物还可以合其他镇痛药物联用达到协同镇痛的作用。原人参二醇类化合物通过特异性刺激强啡肽a的表达,从而达到镇痛的作用。
106.术语
107.除非另有定义,否则本文中所用的全部技术术语和科学术语均具有如本发明所属领域普通技术人员通常理解的相同含义。
108.如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
109.如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由
…
构成”、或“由
…
构成”。
110.在本发明中,术语“躯体依赖(physical dependence)”、“身体依赖”和“生理依赖”可互换使用,指一旦中断使用成瘾药物,可引发戒断综合征的依赖性。
111.在本发明中,术语“戒断综合征”指由于连续使用成瘾物质,从而使产生依赖性的患者一旦中断使用,身体所产生的剧烈生理反应而出现的一系列症状,如出汗、流泪、打哈欠、寒战、起鸡皮疙瘩、瞳孔散大、呕吐、腹泻、腹痛、心律增加、血压上升、失眠、震颤等综合症状。
112.在本发明中,术语“精神依赖(psychological dependence)”与“心理依赖”可互换使用,是指患者对药物的渴求,以期获得服用成瘾药物后的特殊快感。
113.通过“治疗(treating)”或“治疗(treatment)”和/或“预防(preventing)”或“预防(prevention)”,作为一个整体,是指至少抑制或改善影响个体的相关症状,其中,抑制和改善使用其广义,指至少降低参数的量级,所述参数如所治疗的病症相关的症状,如疼痛。因此,本发明的方法包括预防和处理各种不同的疼痛。
114.疼痛
115.在本发明提供将本发明活性成分或其制剂用于治疗疼痛的应用。
116.在本发明中,疼痛没有特别限制,代表性的例子包括(但并不限于)偏头痛、背痛、颈痛、妇科疼痛、分娩前或分娩疼痛、矫形疼痛、中风后疼痛、外科手术后或操作性疼痛、疱疹后神经痛、镰状细胞危象、间质性膀胱炎、泌尿疼痛(如尿道炎)、牙齿疼痛、头痛、伤口或
如外科手术(如囊炎切除术或臀部、膝盖或其他关节置换术)的医疗操作产生的疼痛、缝合、骨折复位、活检等。疼痛还可出现在患有癌症的患者中,这可能由多方面原因造成,如炎症、神经压迫和因肿瘤入侵和肿瘤转移到骨骼或其他组织而产生的组织肿胀所造成的机械力。
117.在另一优选例中,所述的疼痛包括(但并不限于):周围神经性疼痛、中枢神经性疼痛、异常性疼痛、灼性神经痛、痛觉过敏、感觉过敏、痛觉过度、神经痛、神经炎和神经病变。
118.成瘾
119.药物成瘾和药物依赖是一种慢性复发性脑部疾病,主要表现为对成瘾药物的强迫用药行为和药量的不可控性。出现物质依赖状况后,若突然停止服用药物,可能出现药物戒断症状。许多原本用于医学用途的药品,可能造成物质依赖现象;成瘾物质若是被法律管制,视为非法者,则被称为毒品。这些成瘾物质包括阿片类药物和海洛因、冰毒(甲基苯丙胺)、可卡因、大麻、酒精和尼古丁等。
120.甲基苯丙胺,俗称冰毒,是一种高度成瘾的兴奋剂,是全球最常用的第二大非法药物。滥用甲基苯丙胺或其他安非他明类兴奋剂已成为公共卫生的重要问题。相对于海洛因、可卡因等传统毒品而言,甲基苯丙胺合成工艺简单、前体廉价易得,对中枢神经系统的兴奋作用更强,形成成瘾所需用药次数或累计用药量更少,对吸毒者身体造成的损害更为严重。
121.酒精是一种具有高度成瘾特性的精神活性物质。全球酒精依赖患者达到1.4亿,其滥用和依赖给个人和社会带来了严重的不良影响和经济负担。全世界每年约330万人因过度使用酒精导致死亡。酒精的有害使用还会导致酒精肝,肝硬化等疾病。酒精滥用,酒精成瘾已经成为严峻的公共卫生灾难和危害人类健康的世界性难题,是仅次于心血管疾病和肿瘤之后,位居第三位的全球公共卫生问题。
122.尼古丁,又名烟碱,是一种强效拟副交感神经生物碱,是香烟内的主要有效成分。尼古丁依赖是吸烟者的主要特征,它指个体在反复使用尼古丁后导致生理和心理变化,包括使用渴求增强且难以控制,不计危害后果而持续且优先地使用,耐受性增加且出现戒断症状。烟草依赖是目前非常严重的公共卫生问题之一。who指出,烟草每年使700多万人失去生命,其中有600多万人源于直接使用烟草,约89万人是接触二手烟的非吸烟者。
123.反复使用阿片类药物包括吗啡和芬太尼,机体会产生镇痛耐受,必须增加剂量才能获得相同的镇痛效果。长期或反复使用阿片类药物也可导致成瘾,包括躯体依赖(生理依赖)和精神依赖(心理依赖)两个方面。躯体依赖性是为避免戒断症状而反复用药,且由于耐受性剂量逐渐加大,在成瘾过程中表现为厌恶效应,起到负强化效应。精神依赖性是指依赖者的心理觅药的渴求和重复用药达到的欣快感,表现为奖赏作用,起到正强化效应,促使患者屡次复吸。
124.活性成分
125.如本文多用,“本发明的活性成分”、“本发明的活性化合物”可互换使用,均指原人参二醇类化合物,包括原人参二醇、原人参三醇。其中,原人参二醇包括20(s)-原人参二醇、20(r)-原人参二醇、或其组合(如外消旋物)。所述的原人参三醇包括20(s)-原人参三醇、20(r)-原人参三醇、或其组合(如外消旋物)。此外,所述术语包括天然产物或人工合成或修饰的产物。
126.应理解,本发明的活性成分包括本发明的活性化合物(原人参二醇、原人参三醇、或其组合)、或其药学上可接受的盐或酯、对映异构体、非对映异构体或外消旋体、或其前
药。应理解,本发明的活性成分还包括本发明的活性化合物的晶型、无定形化合物、溶剂化物、水合物等形式。
127.所述“药学上可接受的盐(或酯)”为本发明的活性化合物与无机酸或有机酸反应形成常规的无毒盐(或酯)。例如,常规的无毒盐可通过本发明的活性化合物与无机酸或有机酸反应制得,所述无机酸包括盐酸、氢溴酸、硫酸、硝酸、胺基磺酸和磷酸等,所述有机酸包括柠檬酸、酒石酸、乳酸、丙酮酸、乙酸、苯磺酸、对甲苯磺酸、甲磺酸、萘磺酸、乙磺酸、萘二磺酸、马来酸、苹果酸、丙二酸、富马酸、琥珀酸、丙酸、草酸、三氟乙酸、硬酯酸、扑酸、羟基马来酸、苯乙酸、苯甲酸、水杨酸、谷氨酸、抗坏血酸、对胺基苯磺酸、2-乙酰氧基苯甲酸和羟乙磺酸等;或者本发明的活性化合物与丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、天冬氨酸或谷氨酸形成酯后再与无机碱形成的钠盐、钾盐、钙盐、铝盐或铵盐;或者本发明的活性化合物与赖氨酸、精氨酸、鸟氨酸形成酯后再与盐酸、氢溴酸、氢氟酸、硫酸、硝酸或磷酸形成的对应的无机酸盐或与甲酸、乙酸、苦味酸、甲磺酸或乙磺酸形成的对应的有机酸盐。
128.其他镇痛类药物
129.1)阿片类镇痛药包括可待因、双氢可待因、吗啡、芬太尼、舒芬太尼、瑞芬太尼、哌替啶、羟考酮;
130.2)抗癫痫药物包括卡马西平、苯妥英钠和加巴喷丁类药物如加巴喷丁、普瑞巴林和米罗巴林;
131.3)非甾体抗炎镇痛药物包括氟比洛芬酯、布洛芬、双氯芬酸钠、美洛昔康、萘普生、塞来昔布和普瑞昔布;
132.4)单胺类神经递质重摄取抑制剂抗抑郁药包括阿米替林和度洛西丁;
133.5)局部麻醉药包括利多卡因、罗哌卡因、丙胺卡因;
134.6)去甲肾上腺素α2受体激动剂如可乐定和右美托咪定;
135.7)mor-nri双靶点镇痛药如地佐辛、他喷他多、喷他佐辛和曲马多;
136.8)抗偏头痛药如cgrp抗体及其受体拮抗剂;
137.9)中草药包括独一味提取物及其有效成分如山栀子苷甲酯和8-o-乙酰山栀子苷甲酯、乌头/附子及其有效成分如草乌甲素、高乌甲素和雪上一支蒿甲素,以及延胡索及其有效成分如四氢巴马汀、千金藤啶碱、紫堇达明和脱氢紫堇鳞茎碱。
138.药物组合物及施用方法
139.本发明还提供了含有本发明活性成分的组合物或制剂或产品,所述组合物或制剂或产品可用于抗衰老。代表性的组合物或制剂或产品包括抗衰老的药物、保健品、和化妆品。
140.一种优选的组合物是药物组合物,它含有有效量的维拉帕米或其药学上可接受的盐和药学上可接受的载体。
141.如本文所用,术语“有效量”或“有效剂量”是指可对人和/或动物产生功能或活性(即抗衰老功能)的且可被人和/或动物所接受的量。
142.如本文所用,术语“药学上可接受的”的成分是适用于人和/或哺乳动物而无过度不良副反应(如毒性、刺激和变态反应)的,即具有合理的效益/风险比的物质。术语“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。
143.本发明的药物组合物含有安全有效量的本发明的活性成分以及药学上可接受的载体。这类载体包括(但并不限于):盐水、缓冲液、葡萄糖、水、甘油、乙醇、及其组合。通常药物制剂应与给药方式相匹配,本发明的药物组合物的剂型为注射剂、口服制剂(片剂、胶囊、口服液)、透皮剂、缓释剂。例如用生理盐水或含有葡萄糖和其他辅剂的水溶液通过常规方法进行制备。所述的药物组合物宜在无菌条件下制造。
144.本发明所述的活性成分的有效量可随给药的模式和待治疗的疾病的严重程度等而变化。优选的有效量的选择可以由本领域普通技术人员根据各种因素来确定(例如通过临床试验)。所述的因素包括但不限于:所述的活性成分的药代动力学参数例如生物利用率、代谢、半衰期等;患者所要治疗的疾病的严重程度、患者的体重、患者的免疫状况、给药的途径等。通常,当本发明的活性成分每天以约0.001-100mg/kg动物体重(较佳的0.01-50mg/kg,更佳地0.05-20mg/kg动物体重)的剂量给予,能得到令人满意的效果。例如,由治疗状况的迫切要求,可每天给予若干次分开的剂量,或将剂量按比例地减少。
145.典型地,当以口服方式给予本发明的活性成分,优选地,施用对象为人时,口服剂量可以为0.05-50mg/kg,较佳地0.10-20mg/kg。
146.本发明的主要优点包括:
147.a)本发明的活性成分在有效镇痛的同时,不产生镇痛耐受性、躯体依赖性、精神依赖性和成瘾。
148.b)本发明的活性成分有效镇痛的同时,可以治疗或缓解阿片类药物诱导的成瘾包括躯体以及精神依赖性。
149.下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如(sambrook等,分子克隆:实验室手册,new york:cold spring harbor laboratory press,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
150.实验通用方法
151.1大鼠鞘内置管:
152.大鼠在呼吸麻醉机下用5%异氟烷快速麻醉(麻醉机气流速度为0.3l/min),随后由2%异氟烷维持麻醉。将18-cm的聚乙烯导管(pe-10:外径:0.55mm,内径:0.3mm)从大鼠腰部水平处沿脊柱处插入。在进行药物治疗前一周,用10μl 4%利多卡因检测插管情况,蛛网膜下腔鞘内注射若大鼠出现双侧后足瘫软,且恢复后无运动障碍,说明插管成功可进行后续实验。
153.2大鼠福尔马林疼痛模型:
154.实验前将大鼠在尺寸为23
×
35
×
19cm的透明观察笼中适应30分钟。取出大鼠左侧足部皮下注射5%的福尔马林溶液(50μl),注射后大鼠立刻放入观察笼,在注射后每隔10分钟测定60秒内的抬脚次数,直到90分钟截止。
155.3大鼠脊神经结扎神经病理性疼痛模型:
156.大鼠在呼吸麻醉机下用5%异氟烷快速麻醉(麻醉机气流速度为0.3l/min),随后由2%异氟烷维持麻醉。在腰脊髓部钝性分离左侧肌肉,暴露和去除l6横突,暴露l5神经并用6-0丝线扎紧;去除骶骨夹角下方附近筋膜,挑出l6神经并用6-0丝线扎紧。术后大鼠单笼
饲养恢复一周。用von frey电子测痛仪测定后足底机械痛阈值,<8g且无运动障碍视为造模成功,用于后续实验。
157.4大鼠骨癌疼痛模型:
158.雌性大鼠按50mg/kg腹腔注射戊巴比妥钠进行麻醉,在大鼠胫骨处开一0.5-cm切口,钝性分离肌肉后暴露胫骨,使用5号针头在胫骨中间轻轻钻孔。随后微量注射器注射10μl walker 256乳腺癌细胞(4
×
105个/ml),注射完毕后停针30秒,拔出针头立刻用无菌骨蜡封住伤口,随后消毒、缝合和放入笼中。两周后测定足机械痛阈值,<8g大鼠视为造模成功,用于后续实验。
159.5大鼠炎性疼痛模型:
160.异氟烷麻醉动物,在左后爪的胫跗骨关节中缓慢注入100μl cfa。cfa注射两天后测定机械痛和热痛阈值。
161.6大鼠机械痛和热痛阈值测定:
162.将大鼠分别置于机械痛和热痛检测架上。对于大鼠机械痛测定,用电子机械痛阈检测仪垂直刺激大鼠后肢足底中部,检测仪安装了15号纤维,测量时,缓慢增加刺激力度,直至纤维弯曲成s形,持续6-8秒,观察大鼠是否出现缩足或抬脚反应。记录大鼠缩足或抬脚的最小阈值作为足收缩阈值(paw withdrawal threshold,pwt)。每隔3分钟检测一次,重复三次,三次的平均值作为大鼠该足的机械疼痛阈值。机械疼痛阈值反映大鼠机械刺激伤害/疼痛程度。对于大鼠热痛测定,打开垂直放置于大鼠后肢脚掌处的玻璃板下方的辐射热源进行检测,观察大鼠足底开始接受辐射热源到突然舔足,抬足或缩足的总时长为其受辐射热刺激后痛觉阈值,用缩足反应潜伏期(paw withdrawal latency,pwl)表示。以30秒为最大测量阈值,每隔5分钟检测一次,重复三次,三次的平均值作为大鼠该足辐射热痛阈值。辐射热疼痛阈值反映大鼠热刺激伤害/疼痛程度。
163.7小鼠条件位置偏好(conditioned place preference,cpp)模型建立:
164.为期10天的cpp模型包括三个阶段:前测试期,获得期和后测试期。前测试期(1-4天):允许雄性swiss小鼠在三个隔室自由穿梭,每天2次,每次15分钟,共3天。在第4天,记录小鼠自由穿梭于三个隔室15分钟内每个隔室的停留时间。获得期(5-9天):小鼠每隔6小时(上午9:00和下午3:00)进行皮下交替注射5天吗啡(10mg/kg)或生理盐水(10ml/kg),接着立刻放入隔室内,训练45分钟。在获得期的第5、7和9天,分别上午9:00皮下注射吗啡(10mg/kg)和下午3:00皮下注射生理盐水(10ml/kg),并放入吗啡伴药箱中和生理盐水配对箱中训练45分钟。在第6和8天,交换吗啡和生理盐水的注射时间。后测试期(第10天):允许小鼠自由进出三个隔室15分钟进行测试。条件位置偏好得分由小鼠在伴药箱中停留的时间减去在生理盐水配对箱中所花费的时间。本实验由3ccd相机拍摄小鼠在每个隔室的穿梭活动,并使用ethovision xt 8.0软件记录小鼠在每个隔室停留的时间。
165.8原代细胞培养:
166.出生24小时内任一性别的wistar大鼠分离获得脊髓,剥膜剪碎后加入0.05%胰酶在5%二氧化碳培养箱中消化7-9分钟。离心去上清,将离心管内底部消化后的组织用10%fbs、1%双抗的dmem培养基吹散重悬,依次70-和40-μm筛网过滤,接种至多聚赖氨酸(0.1mg/ml)包被的75cm2培养瓶中,放入5%二氧化碳培养箱培养10天。
167.制备小胶质细胞时,培养瓶放入摇床在37℃摇荡(260rpm)1.5-2小时,收集细胞悬
浮液、离心、重悬细胞,后接种于新的细胞培养板中,次日用预温的pbs洗去未黏附细胞。通过小胶质细胞标记蛋白iba-1的免疫荧光测定,获得的小胶质细胞纯度大于95%。
168.制备星形胶质细胞时,培养的细胞弃去培养基,经pbs洗两遍后加入0.05%含edta的胰酶。在37℃条件下消化3分钟除去少突胶质细胞,终止消化并去除细胞悬浮液,剩下的贴壁单层星形胶质细胞继续用胰酶消化传代以供后续使用。通过星形胶质细胞标记蛋白gfap的免疫荧光测定,获得的星形胶质细胞纯度大于90%。
169.制备神经元细胞时,细胞悬浮液经40-μm筛网过滤后,接种于10-cm的细胞培养皿中,放入细胞培养箱培养30分钟。然后吸取未贴壁的上层细胞悬液,将其接种于多聚赖氨酸培养板中。培养1.5-2小时后,将dmem更换成含1
×
b27神经营养因子及0.5mm谷氨酰胺的neurobasal培养基,继续培养3-4天。通过神经元细胞标记蛋白neun的免疫荧光测定,获得的神经元细胞纯度大于85%。
170.9细胞及组织总rna提取及实时定量pcr测定:
171.戊巴比妥钠(50mg/kg,i.p.)麻醉大鼠用,迅速断头放血,取出脊髓腰膨大(l3-l5)组织,按比例加入trizol试剂(50mg/ml)匀浆以萃取、沉淀rna,最后根据rna沉淀量加入适量depc水使其溶解。采用微量酶标仪测定提取rna的浓度及纯度。
172.采用逆转录试剂盒在普通pcr仪上运行相应的程序,将提取的总rna逆转录成cdna后-20℃保存备用。后续的实时定量pcr操作采用sybr qpcr mix,检测强啡肽前体基因(pdyn),内啡肽前体基因(pomc),脑啡肽前体基因(pnoc),和nociceptin/orphaninfq前体基因(penk)ct值,以gapdh作为内参基因,采用2-δδct
方法计算目标基因相对表达量。
173.10强啡肽a和β-内啡肽蛋白质含量测定:
174.大鼠取出脊髓腰膨大处(l3-l5)组织,用10mm tris-hcl(5ml/1g组织)均浆化(4,000rpm,15秒),并在4℃条件下离心(5000rpm)15分钟后获得的上清液。此外,新生大鼠脊髓来源的原代细胞给药处理培养2小时后收集细胞培养上清液。根据酶联免疫试剂盒说明书,测定细胞培养和脊髓组织上清液中强啡肽a和β-内啡肽含量。
175.11组织免疫荧光染色:
176.大鼠腹腔注射戊巴比妥钠(50mg/kg)麻醉,沿胸骨剑突下缘打开胸腔,暴露和游离心脏。迅速经左心室将针头插入主动脉,用4-0号手术缝合丝线固定针头,剪开右心耳。缓慢灌注100ml生理盐水将血液冲洗后,继续灌注60ml 4%甲醛溶液。然后取出脊髓腰膨大部分(l3-l5)置于4%甲醛固定液4℃过夜,然后依次经蔗糖溶液梯度脱水、包埋、冰冻切片(厚度为30μm)和-20℃保存备用。冻存的组织切片复温后用封闭液室温封闭1小时,随后使用封闭液配制一抗(强啡肽a抗体、小胶质细胞标记iba-1、星形胶质细胞标记gfap和神经元细胞标记neun)在4℃孵育18-24小时。一抗孵育结束后,加入配制二抗的封闭液在37℃条件下培养1小时,随后用抗荧光淬灭封片剂封片,-20℃避光保存备用。使用leica tcs sp8激光共聚焦显微镜拍摄成像,并用image j图像处理软件进行荧光定量和荧光染色共定位分析。
177.实施例1口服20(s)-原人参二醇在神经病理性疼痛模型大鼠模型中镇痛作用
178.方法
179.选取l5/l6脊神经结扎的神经病理性疼痛大鼠,随机分为六组(每组6只)。其中一组口服溶剂(乙醇:丙烯乙二醇:蒸馏水=2:7:1比例,6.5ml/kg),另外5组分别口服不同剂量的20(s)-原人参二醇(五组口服ppd剂量分别为1、3、10、30或100mg/kg)。在给药前及给药
后0.5、1、2、4小时不同时间点测定大鼠对机械刺激和热辐射刺激的缩足反应阈值或缩足潜伏时间(热辐射刺激在机械刺激10分钟后进行)。在1小时测得各计量组机械痛阈值和热辐射痛阈值以计算%最大可能效应(%maximal possible effect,%mpe),然后进行剂量-反应分析。
180.结果
181.在4小时的检测时间内,生理盐水组大鼠健侧和手术患侧的机械痛阈值及热辐射痛阈值基本不变,而口服20(s)-原人参二醇可剂量依赖性地抑制大鼠手术患侧的机械痛敏(图1a和1b)和热辐射痛敏(图1c和1d),但不影响健侧疼痛阈值。镇痛作用可持续约3小时,镇痛效果最强时间点为1小时。
182.由图1b和1d的剂量-反应分析结果显示,口服20(s)-原人参二醇在神经结扎引起的机械痛敏和热辐射痛敏中的最大镇痛效应分别为e
max
为61%和68%mpe,ed
50
分别为5.8和4.5mg/kg。
183.综上,口服20(s)-原人参二醇可以抑制神经病理性疼痛,且这种抑制作用的程度和服用的20(s)-原人参二醇剂量呈正向依赖性。并且,口服20(s)-原人参二醇镇痛效果也非常明显。
184.实施例2口服20(s)-原人参二醇在不同病因引起的疼痛大鼠模型中的镇痛作用。
185.方法
186.为验证20(s)-原人参二醇对其他不同病因疼痛模型的镇痛作用,我们采用了骨癌疼痛模型、cfa引发的炎性疼痛模型和福尔马林诱发疼痛模型。
187.两组骨癌疼痛大鼠(每组6只)和两组cfa引发的炎性疼痛大鼠(每组6只)分别口服溶剂(6.5ml/kg)或20(s)-原人参二醇(100mg/kg),在给药前及给药后0.5、1、2、4小时不同时间点测定大鼠对机械刺激和热辐射刺激的缩足反应阈值或缩足潜伏时间(热辐射刺激在机械刺激10分钟后进行)。
188.此外,两组大鼠(每组6只)在足底注射5%福尔马林前30分钟口服溶剂(6.5ml/kg)或20(s)-原人参二醇(100mg/kg)。
189.结果
190.口服20(s)-原人参二醇可以显著缓解大鼠骨癌疼痛(图2a和2b)和cfa引发的炎性疼痛(图2c和2d)。
191.福尔马林可以引起大鼠i相和ii相舔足反应。口服20(s)-原人参二醇可以抑制大鼠福尔马林引起的ii相舔足次数,但对i相疼痛并无影响(图2e)。
192.实施例3口服20(s)-原人参二醇联合加巴喷丁或吗啡的协同镇痛效应
193.方法
194.为验证20(s)-原人参二醇与加巴喷丁或吗啡之间的相互作用,给予最小有效剂量的20(s)-原人参二醇(3mg/kg),加巴喷丁(10mg/kg)和吗啡(0.3mg/kg)。第一个实验四组大鼠(每组6只),每组分别服用溶剂(6.5ml/kg) 生理盐水(3ml/kg),20(s)-原人参二醇(3mg/kg) 生理盐水(3ml/kg),溶剂(6.5ml/kg) 加巴喷丁(10mg/kg)和20(s)-原人参二醇(3mg/kg) 加巴喷丁(10mg/kg)。
195.第二个实验四组大鼠(每组6只),每组分别服用溶剂(6.5ml/kg) 生理盐水(3ml/kg),20(s)-原人参二醇(3mg/kg) 生理盐水(3ml/kg),溶剂(6.5ml/kg) 吗啡(0.3mg/kg)和
20(s)-原人参二醇(3mg/kg) 吗啡(0.3mg/kg)。
196.在给药前及给药后0.5、1、2、4小时不同时间点测定各大鼠对机械刺激和热辐射刺激的缩足反应阈值或缩足潜伏时间(热辐射刺激在机械刺激10分钟后进行)。
197.结果
198.单独给予小剂量20(s)-原人参二醇或加巴喷丁在给药后1小时抗机械痛敏作用分别为20%mpe和22%mpe,抗热痛敏作用分别为20%mpe和30%mpe,在该剂量下镇痛效果较弱。但巴喷丁和20(s)-原人参二醇联合给药,相比任一单独给药,均显著增强抗机械痛敏达到55%mpe,热痛敏作用增强达到62.2%mpe。采用金氏公式(q=(e
a b
)/(ea e
b-ea*eb)计算(金正均,中国药理学报,1:70-76,1980;金正均、张效文,上海第二医学院学报,1:15-18,1981),q值分别为1.46和1.40,均大于1.15。这表明加巴喷丁和20(s)-原人参二醇联合给药,显著增强缓解大鼠机械痛敏和热辐射痛敏作用,表现为协同镇痛效应(图3a和3b)。
199.同样,小剂量吗啡或20(s)-原人参二醇单独给药,产生较弱的镇痛作用,在给药1小时后其抗机械痛敏作用分别为20%mpe和25%mpe,抗热痛敏作用分别为20%mpe和27%mpe。二者联合使用,抗机械痛敏作用增加到54%mpe,热辐射痛敏作用增加到74%mpe。采用金氏公式计算,它们的q值分别为1.35和1.78,均大于1.15。这表明二药联合使用产生显著的协同镇痛作用(图3c和3d)。
200.实施例4口服20(s)-原人参二醇对大鼠脊髓小胶质细胞表达强啡肽a的特异性刺激作用
201.方法
202.采用两组l5/l6脊神经结扎的神经病理性疼痛大鼠(每组6只),分别口服溶剂(6.5ml/kg)或20(s)-原人参二醇(100mg/kg),给药一小时后断头并取腰椎膨大处(l3-l5)手术侧脊髓组织。通过实时定量pcr测定pdyn,pomc,penk和pnoc基因表达水平,结果如图4a所示。同时通过酶联免疫荧光法测定脊髓匀浆上清液强啡肽a和β-内啡肽水平。结果如图4b所示。另外采用两组假手术大鼠口服生理盐水(6.5ml/kg)或20(s)-原人参二醇(100mg/kg),给药一小时后断头并取腰椎膨大处(l3-l5)手术侧脊髓组织,以测定阿片样肽基因和蛋白表达,结果如图4c和4d所示。
203.选用两组神经病理性疼痛大鼠,采用免疫荧光染色法对脊髓切片进行共染实验:强啡肽a与小胶质细胞标记蛋白iba-1、星形胶质细胞标记蛋白gfap或神经元细胞标记蛋白neun的共染图像如图5a-5n所示。
204.进一步,用不同浓度的20(s)-原人参二醇(1、3、10、30和100μm)处理原代脊髓小胶质细胞,培养2小时后,检测小胶质细胞强啡肽a基因和蛋白表达,如图6a和6b所示;用20(s)-原人参二醇(100μm)处理原代脊髓星形胶质细胞和神经元细胞,培养2小时后,检测星形胶质细胞和神经元细胞强啡肽a基因和蛋白表达,如图6c和6d所示。
205.结果
206.由图4a可知,在脊神经结扎的神经病理性疼痛的大鼠中,服用20(s)-原人参二醇使得脊髓pdyn表特异性升高达,而不影响pomc,penk和pnoc表达。且明服用20(s)-原人参二醇可以特异性刺激脊髓释放强啡肽a,而不影响β-内啡肽释放(图4b)。
207.由图4c可知,在假手术大鼠模型中,口服20(s)-原人参二醇也显著增加脊髓pdyn基因表达,而不增加pomc,penk或pnoc基因表达水平。与此同时,口服20(s)-原人参二醇显
著增加脊髓强啡肽a蛋白表达,而不影响β-内啡肽蛋白表达(图4d)
208.由图5a-5d可知,强啡肽a与小胶质细胞iba-1在脊髓有免疫荧光共表达,口服20(s)-原人参二醇明显增加脊髓强啡肽a和iba-1共表达。通过imagej定量分析,给药组的共染区域面积为对照组的2.1倍(图5e)。
209.由图5f-5i和5k-5n可知,同样在大鼠脊髓,强啡肽a与星形胶质细胞标记蛋白gfap或神经元细胞标记蛋白neun均有共表达。
210.由图5j可知,与服用生理盐水(对照组)相比,口服20(s)-原人参二醇(给药组)的强啡肽a与gfap的共染面积没有明显改变。
211.同样地,由图5o可知,口服20(s)-原人参二醇不明显改变强啡肽a与neun的共染面积。
212.如图6a和6b可知,20(s)-原人参二醇处理呈剂量依赖性地增加脊髓小胶质细胞表达强啡肽a基因和蛋白质,ed
50
值分别为13和19.8μm。而由图6c和6d可知,20(s)-原人参二醇不明显改变脊髓星形胶质细胞或神经元表达强啡肽a基因或蛋白。
213.实施例5蛛网膜下腔鞘内预先给予小胶质活化抑制剂米诺环素、强啡肽a抗血清和特异性κ-阿片受体拮抗剂对20(s)-原人参二醇镇痛的抑制作用
214.方法
215.采用两组神经病理性疼痛大鼠(每组6只),分别蛛网膜下腔鞘内预先注射生理盐水(10μl)或小胶质细胞活化抑制剂米诺环素(minocycline,100μg)。4小时后,两组均口服20(s)-原人参二醇(100mg/kg)。第一次给药前,第二次给药前及给药后0.5、1、2和4小时测定大鼠后足对机械刺激的缩足反应阈值和热辐射的缩足反应潜伏期,结果如图7a、7b所示。
216.采用三组神经病理性疼痛大鼠(每组6只),分别蛛网膜下腔鞘内预先注射空白兔血清(10μl)、强啡肽a抗血清(1:10,10μl)或β-内啡肽抗血清(1:10,10μl)。0.5小时后,三组均口服20(s)-原人参二醇(100mg/kg)。测定大鼠后足对机械刺激的缩足反应阈值和热辐射的缩足反应潜伏期,结果如图8a、8b所示。
217.强啡肽a是内源性κ-阿片受体激动剂,为验证20(s)-原人参二醇镇痛作用是否通过激动κ-阿片受体达到,作以下验证:采用四组神经病理性疼痛大鼠(每组6只)分别蛛网膜下腔鞘内注射生理盐水(10μl)、μ-阿片受体拮抗剂ctap(10μg)、κ-阿片受体拮抗剂gnti(50μg)或δ-阿片受体拮抗剂naltrindole(5μg)。0.5小时后,四组大鼠均口服20(s)-原人参二醇(100mg/kg),结果如图8c和8d所示。
218.结果
219.由图7a、7b可知,口服20(s)-原人参二醇产生时间依赖式的镇痛作用,米诺环素不影响疼痛的基础阈值,但完全抑制20(s)-原人参二醇所产生的镇痛作用。
220.由图8a、8b可知,口服20(s)-原人参二醇产生时间依赖式的镇痛作用,强啡肽a抗血清不影响疼痛的基础阈值,但完全抑制20(s)-原人参二醇所产生的镇痛作用,而β-内啡肽抗血清不能阻断20(s)-原人参二醇产生的镇痛作用。
221.由图8c、8d可知,口服20(s)-原人参二醇(100mg/kg)产生时间依赖式的镇痛作用,gnti不影响疼痛的基础阈值,但完全抑制20(s)-原人参二醇产生的镇痛作用,而ctap或naltrindole未能阻断20(s)-原人参二醇所产生的镇痛作用。
222.实施例6口服20(s)-原人参二醇对吗啡镇痛耐受和躯体依赖的抑制作用
223.采用四组神经病理性疼痛大鼠(每组6只),每一组分别连续每日两次给予溶剂(6.5ml/kg) 生理盐水(1ml/kg),20(s)-原人参二醇(30mg/kg) 生理盐水(1ml/kg),溶剂(6.5ml/kg) 吗啡(3mg/kg)或20(s)-原人参二醇(30mg/kg) 吗啡(3mg/kg),连续7天。每次早晨给药1小时后,测定大鼠后足对机械刺激的缩足反应阈值和热辐射的缩足反应潜伏期,结果如图9a和9b所示。
224.在第8天早晨,四组大鼠均口服20(s)-原人参二醇(30mg/kg),给药前及给药后0.5、1、2和4小时测定大鼠后足对机械刺激的缩足反应阈值和热辐射的缩足反应潜伏期。在给予20(s)-原人参二醇6小时后,四组大鼠均给予吗啡(3mg/kg),测定接下来4小时大鼠后足疼痛阈值。结果如图9c和9d所示。
225.对上述大鼠再持续口服给药3日,在最后一日早晨给药后4小时,大鼠腹腔注射纳洛酮(5mg/kg),随后立即观察30分钟内大鼠戒断症状,结果如图10a-10e所示。
226.由图9a和9b可知,20(s)-原人参二醇在7天内的镇痛作用保持不变,而吗啡镇痛作用在7天内逐渐产生耐受并最终完全消失。与单独使用20(s)-原人参二醇或吗啡相比,同时给予20(s)-原人参二醇和吗啡不仅产生明显的镇痛协同作用,而且还能够完全抑制吗啡镇痛耐受反应。
227.由图9c和9d可知,对于分别连续一周给予生理盐水、20(s)-原人参二醇、吗啡、20(s)-原人参二醇 吗啡联合的四组大鼠,再单剂量口服20(s)-原人参二醇后,大鼠均产生显著的时间依赖式镇痛作用。此外,单剂量皮下注射吗啡对于连续一周吗啡给药从而产生吗啡耐受的大鼠,不能产生镇痛作用;但对于连续一周生理盐水给药组、20(s)-原人参二醇给药组和20(s)-原人参二醇 吗啡联合给药组,再进行单剂量皮下注射吗啡能产生显著的镇痛作用。
228.图10a-10e表明,与生理盐水对照组比较,口服20(s)-原人参二醇不产生躯体依赖性,而吗啡产生明显的躯体依赖性,联合应用20(s)-原人参二醇(30mg/kg)显著减少吗啡相关的戒断症状,包括颤抖(图10a),跳跃(图10b),牙齿颤动(图10c),腹泻(图10d)和湿狗样抖动(图10e)。
229.实施例7口服20(s)-原人参二醇对吗啡条件性位置偏好(cpp)获得的抑制作用
230.两组小鼠(每组10只)每天交替口服溶剂(10ml/kg)或20(s)-原人参二醇(100mg/kg),连续5日,随后进行条件位置偏好测试,结果如图11a所示。
231.另外四组小鼠(每组10只)每天皮下注射生理盐水(10ml/kg)或吗啡(10mg/kg)5天,在最后一次注射前50分钟,小鼠单剂口服溶剂(10ml/kg)或20(s)-原人参二醇(100mg/kg),随后立即进行15分钟位置偏好测试,如图11b所示。
232.由图11a可知,两组小鼠在前测试期和后测试期均未获得条件位置偏好,表明长期口服20(s)-原人参二醇不产生条件位置偏好。
233.由图11b可知,在前测试期,四组小鼠均未显示条件位置偏好。在后测试期,生理盐水组未出现条件位置偏好,但皮下注射吗啡组显示出明显的条件位置偏好。单剂口服20(s)-原人参二醇(100mg/kg)不影响生理盐水组条件位置偏好反应,但完全阻断吗啡诱导的条件位置偏好的获得。
234.实施例8蛛网膜下腔鞘内预先给予糖皮质激素受体拮抗剂对20(s)-原人参二醇镇痛的抑制作用
mesylate(100nm)0.5小时后,分别给与20(s)-原人参二醇(100μm)、特异性糖皮质激素受体激动剂dex(100nm)或不透过细胞膜的dex与牛血清白蛋白(bsa)的缀合物dex-bsa(10nm)并培养2小时,随后检测小胶质细胞强啡肽a基因和蛋白表达。如图14a和14b可知,20(s)-原人参二醇、dex和dex-bsa显著增强小胶质细胞强啡肽a基因表达;而dex 21-melylate不影响小胶质细胞强啡肽基因的基础表达,但完全抑制20(s)-原人参二醇、dex和dex-bsa对强啡肽a基因表达的刺激作用。而由图14b可知,dex-21-melylate不影响小胶质细胞的强啡肽a蛋白基础表达,但完全抑制20(s)-原人参二醇、dex和dex-bsa对强啡肽a蛋白表达的刺激作用。
242.上述结果说明,20(s)-原人参二醇通过激动脊髓小胶质细胞糖皮质激素受体(可能是细胞膜糖皮质激素受体),刺激小胶质细胞表达和释放强啡肽a,从而产生镇痛作用。
243.讨论
244.20(s)-原人参二醇的生物活性已有一些报道,但本发明之前目前未报道过20(s)-原人参二醇具有治疗疼痛的用途,也未公开过可用于治疗阿片类药物(或成瘾物质)的躯体依赖和精神依赖。本发明提供,20(s)-原人参二醇在大鼠/小鼠神经病理性疼痛、癌症疼痛、炎性疼痛和福尔马林疼痛等模型有显著的镇痛作用;20(s)-原人参二醇长期给药不产生镇痛耐受作用、躯体依赖性和条件性位置偏好作用(精神依赖性);20(s)-原人参二醇有效抑制吗啡诱导的镇痛耐受性、躯体依赖性和精神依耐性。
245.本发明人研究表明,20(s)-原人参二醇的镇痛主要部位在脊髓,20(s)-原人参二醇通过激动脊髓小胶质细胞糖皮质激素受体(可能是细胞膜糖皮质激素受体)极其有效地促进强啡肽a表达和释放,从而出乎意料地产生镇痛作用和戒除治疗阿片类药物(或其他成瘾物质)的躯体依赖和精神依赖等作用。
246.在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。