1.本发明涉及有机合成领域,具体而言,涉及一种非奈利酮中间体的合成方法,此外还涉及非奈利酮的合成方法。
背景技术:
2.非奈利酮化学名称为(4s) 4-(4-氰基-2-甲氧苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺,结构式如下:其是一种非甾体类盐皮质激素受体拮抗剂,被用于治疗慢性肾病。
3.专利文献wo2008104306a、cn106795155a、cn101641352a和cn107849043a均报道了非奈利酮的制备方法。从上述专利申请中不难看出,许多合成步骤需要采用手性柱拆分,手性柱价格昂贵,且在技术上非常费时费力,同时溶剂消耗量大,导致成本高,不适合工业化生产。
4.因此,需要开发具有高收率、高纯度、简便高效、低成本和易于工业化生产的药学活性化合物的制备方法,以满足临床试验和注册申请要求。本发明提供一种非奈利酮中间体的合成方法,该方法具有收率高,后处理简单,成本低,易于工业化的优点,同时使产品具有符合原料药注册所需的纯度。
技术实现要素:
5.本发明的目的之一在于提供一种新的非奈利酮中间体的合成方法,以解决现有的非奈利酮中间体的合成方法存在制备收率低、成本高、后处理复杂及不利于工业化生产的问题。
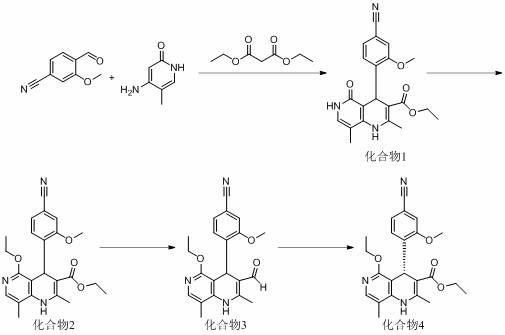
6.为了实现上述目的,本发明第一方面提供了一种新的非奈利酮中间体化合物4的合成方法,该合成方法包括下列步骤:(1)在酸存在下,并在惰性溶剂或无溶剂条件中,由丙二酸二乙酯、4-氰基-2-甲氧基苯甲醛、4-氨基-5-甲基-2-羟基吡啶反应制备得到化合物1;(2)步骤(1)得到的化合物1在惰性溶剂或无溶剂条件中,酸催化下与原甲酸三乙酯发生烷基化反应制备得到化合物2;(3)将步骤(2)得到的化合物2经还原剂还原制备得到化合物3;
(4)在催化剂存在下,化合物3和醇经化学方法拆分,制备得到手性化合物4。
7.工艺路线如下所示:。
8.本发明的第二方面还提供了非奈利酮中间体化合物4,该非奈利酮中间体化合物4由上述合成工艺制备得到。
9.本发明的第三方面还提供了一种新的非奈利酮的合成方法,其合成方法包括下列步骤:将上述化合物4在溶剂中与氨水反应得到非奈利酮i。
10.本发明的第四方面还提供了非奈利酮,该非奈利酮由前述合成工艺制备得到。
11.应用本发明的技术方案,避免使用手性柱拆分方法,降低了成本;所采用化学方法拆分,降低了原料损耗,后处理简单,利于工业化放大生产。
具体实施方式
12.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
13.正如背景技术中所描述的,现有的非奈利酮的合成方法存在制备收率低、成本高、后处理复杂及不利于工业化生产的问题。为了解决上述技术问题,本技术提供了一种新的非奈利酮及其中间体化合物4的合成方法,首先,非奈利酮中间体化合物4的合成方法包括下列步骤:(1) 在酸存在下,并在惰性溶剂或无溶剂条件中,由丙二酸二乙酯、4-氰基-2-甲氧基苯甲醛、4-氨基-5-甲基-2-羟基吡啶反应制备得到化合物1;(2) 步骤(1)得到的化合物1在惰性溶剂或无溶剂条件中,酸催化下与原甲酸三乙酯发生烷基化反应制备得到化合物2;(3) 将步骤(2)得到的化合物2经还原剂还原制备得到化合物3;(4)在催化剂存在下,化合物3和醇经化学方法拆分,制备得到手性化合物4。
14.工艺路线如下所示:
。
15.优选地,步骤(1)和(2)所述的惰性溶剂为醇类溶剂,卤代烃类溶剂,醚类溶剂,芳族烃类溶剂或酰胺类溶剂;所述的醇类溶剂包括但不限于甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇;所述的卤代烃类溶剂包括但不限于二氯甲烷、氯仿、四氯甲烷、1,2-二氯乙烷、三氯乙烷,四氯乙烷、氯苯或氯甲苯;所述的醚类溶剂包括但不限于乙醚、甲基叔丁基醚、四氢呋喃、1,4-二氧六环;所述的芳族烃类溶剂包括但不限于苯、甲苯、二甲苯、硝基苯;所述的酰胺类溶剂包括但不限于n,n-二甲基乙酰胺、n,n-二甲基甲酰胺。
16.优选地,步骤(1)所述的反应在无溶剂条件下进行,反应温度为室温~110℃。所述的酸为醋酸、三氟乙酸、甲烷磺酸或对甲苯磺酸,优选醋酸。
17.优选地,步骤(2)所述的反应在无溶剂条件下进行。反应温度为室温~100℃。步骤(2)所述酸为硫酸,硫酸与化合物1的摩尔比为1:20~1:5,原甲酸三乙酯的量为5~10ml/g 化合物1。
18.优选地,步骤(3)所述的还原剂为硼氢化钠、四氢化铝锂、二异丁基氢化铝四氢呋喃溶液、二异丁基氢化铝甲苯溶液、二异丁基氢化铝环己烷溶液、二异丁基氢化铝庚烷溶液、氢化铝钠四氢呋喃溶液、氢化铝、三叔丁氧基氢化铝锂四氢呋喃溶液或双(2-甲氧乙氧基)氢化铝钠甲苯溶液(60wt%)中的任意一种。
19.优选地,步骤(4)所述的催化剂选自如下结构式所示的化合物中的一种或多种:,,
,,,,,,,,,,,,
,,。
20.所述催化剂与化合物3的摩尔比为0.02~0.5:1。
21.优选地,步骤(4)还包含碱和氧化剂,所述的碱为叔丁醇钠、叔丁醇钾、碳酸铯、氢化钠、碳酸钾、碳酸钠、磷酸钾、4-二甲基氨基吡啶、n,n-二异丙基乙胺、三乙胺、吡啶、咪唑、四丁基氟化铵、2,6-二甲基吡啶、1,8-二氮二环[5.4.0]-7-十一烯中的一种或多种;所述碱与化合物3的摩尔比为2~5:1;所述的氧化剂为4,4'-联苯醌、3,5-二甲基-3',5'-二叔丁基-4,4'-联苯醌、3,5-二甲基-3',5'-二异丙基-4,4'-联苯醌、3,3'-二甲基-5,5'-二叔丁基联苯醌、3,3',5,5'-四叔丁基-[1,1'-双(环己基)]
ꢀ‑
2,2',5,5'-四烯-4,4'-二酮或4-(3,5-二甲基-4-氧代-2,5-环己二烯-1-基)-2,6-二甲基-2,5-环己二烯-1-酮中的一种或多种;所述氧化剂与化合物3的摩尔比为0.5~2:1。
[0022]
上述制备方法中使用的原料和试剂均为现有技术中的已知化合物,可通过商购获得,其中步骤(4)中所述的催化剂也可通过例如公开号为cn101220039a等的中国发明专利申请中记载的合成方法制备而成。
[0023]
本发明的第二方面还提供了非奈利酮中间体化合物4,该非奈利酮中间体化合物4由上述合成工艺制备得到。
[0024]
本发明的第三方面还提供了一种新的非奈利酮的合成方法,该合成方法如下:。
[0025]
具体的合成方法包括下列步骤:将上述化合物4在溶剂中与氨水反应得到非奈利酮i。
[0026]
优选地,所述溶剂为醇,所述的醇类溶剂包括但不限于甲醇、乙醇、正丙醇、异丙醇、正丁醇或叔丁醇;该步骤反应温度为0℃~室温,优选为0℃~10℃,反应时间为10~20小时,优选为16~20小时。
[0027]
本发明的第四方面还提供了非奈利酮,该非奈利酮由前述合成工艺制备得到。
[0028]
应用本发明的技术方案,避免使用手性柱拆分方法,降低了成本;所采用化学方法拆分,降低了原料损耗,后处理简单,利于工业化放大生产。
[0029] 实施例1 化合物1的制备向500ml四口瓶中加入醋酸200ml,35.2g (0.22mol,1.1eq)丙二酸二乙酯,待其溶解后,加入32.2g (0.2mol,1.0eq) 4-氰基-2-甲氧基苯甲醛,室温条件下搅拌30分钟,加入24.8g (0.2mol,1.0eq) 4-氨基-5-甲基-2-羟基吡啶,80℃反应8小时,通过tlc监控反应进程,待反应完全后,将反应液滴加入冰水中,用碳酸氢钠调ph值至7~8,乙酸乙酯萃取,有机相干燥,浓缩,用甲基叔丁基醚打浆得到65.4g化合物1,收率86.2%,纯度97%。
[0030]
lc-ms(m h)
:380.1。
[0031]
实施例2 化合物2的制备将38g化合物1悬浮于200ml的原甲酸三乙酯,升温至130℃,然后,向反应体系中滴加5.4ml浓硫酸(2~3滴/每分钟),滴加完毕后,维持反应温度在130℃条件下搅拌过夜。检测反应完全,冷却,将反应液浓缩至约50ml,加入水300ml,乙酸乙酯500ml萃取,有机相干燥浓缩,用正己烷:乙酸乙酯=10:1打浆纯化,过滤,真空干燥得到37g化合物2,收率91%,纯度98%。
[0032]
lc-ms(m h)
:408.2。
[0033]
实施例3 化合物3的制备在氮气保护下,向500ml四口瓶中加入四氢呋喃150ml,20.4g (0.05mol,1.0eq)化合物2,将反应液冷却到0℃,分批加入2.1g (0.05mol,1.1eq)硼氢化钠,加完后,反应体系温控制在45~50℃,反应2~3小时,点板检测反应完全,用0.5n的稀盐酸淬灭,并用碳酸氢钠调节ph至7~8,加入乙酸乙酯萃取,有机相干燥浓缩,用乙酸乙酯/正己烷=15:1的混合溶剂打浆纯化,得到15.6g化合物3,收率86%,纯度97%。
[0034]
lc-ms(m h)
:364.0。
[0035]
实施例4 化合物4的制备在氮气保护下,向500ml四口瓶中加入15g化合物3(41.3mmol),6.6g催化剂cat. 7(15mmol),16.8g氧化剂 3,5,3',5'-四叔丁基二环己基-2,5,2',5'-四烯-4,4'-二酮(41.3mmol),无水二氯甲烷150ml,然后加入9.5g乙醇(206.5mmol)和6.4g n,n-二异丙基乙胺(49.6mmol), 加完后将反应液加热到40℃反应过夜,用hplc检测反应完全,然后冷却至室温,用硅藻土过滤,滤饼用200ml的二氯甲烷洗涤,滤液在60℃下浓缩除去溶剂,向残留物中加入100ml乙酸乙酯,加热到50℃溶解,再加入10ml正己烷,然后将混合液缓慢降温至10℃(约6h), 在10℃下继续搅拌过夜,过滤,滤饼用冷的乙酸乙酯冲洗3次,得到13.3g化合物4,收率79%,纯度98%,ee值99.5%(手性柱的填充料选择聚(n-甲基丙烯酰基-d-亮氨酸-二环丙基甲基酰胺),流动相为乙酸乙酯)。
[0036]
lc-ms(m h)
:408.2。
[0037]
实施例5 化合物i的制备
向1000ml烧瓶中加入20.4g化合物4,乙醇300 ml,将反应液冷却到0℃,然后向反应体系中滴加氨水(33%,150ml),控制体系温度在5℃以下,加完后继续在5℃以下反应搅拌16小时,有固体析出,点板检测反应完全,过滤,滤饼用冷的水和乙醇洗涤,再用乙酸乙酯和石油醚结晶,将所得溶液搅拌1小时,过滤,在40℃下放入真空干燥箱中干燥,得到16.3g化合物i,收率86.4%,纯度99.5%,ee值 99.4%(手性柱的填充料选择聚(n-甲基丙烯酰基-d-亮氨酸-二环丙基甲基酰胺),流动相为0.5%异丙胺和乙酸乙酯)。
[0038]
lc-ms(m h)
: 379.0。
[0039]1h-nmr (dmso-d6, 400 mhz) δ: 7.71(s, 1h), 7.54(s, 1h), 7.39-7.37(m, 1h), 7.30-7.27(m, 1h), 7.15-7.13 (m, 1h), 6.79-6.75 (m, 2h), 5.39 (s, 1h), 4.06-4.00 (m, 2h), 3.82 (s, 3h), 2.20 (s, 3h), 2.14(s, 3h), 1.07-1.04 (m, 3h)。
[0040]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。