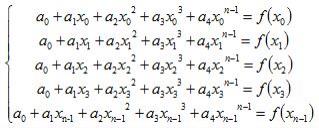
一种清热解毒口服液hplc指纹图谱和在清热解毒口服液质量控制中的应用
技术领域
1.本发明属于药物分析领域,涉及中药质量控制,具体涉及一种清热解毒口服液hplc指纹图谱和在清热解毒口服液质量控制中的应用。
背景技术:
2.清热解毒口服液属清热解毒类中成药,收载于《中国药典》2020年版一部。清热解毒口服液由石膏、金银花、玄参、地黄、黄芩、栀子、连翘等12味中药组成,具有清热解毒功效,用于热毒壅盛所致的发热面赤、烦躁口渴、咽喉肿痛;流感、上呼吸道感染见上述证候者。
3.清热解毒口服液所含中药种类较多,且因为该药使用量较大,生产厂家众多(目前全国有200家以上生产企业),传统质量控制方法难以全面、客观、准确、高效地评价其质量。
4.指纹图谱技术是一种综合的、可量化的质量评价方法,该技术已被广泛应用于中药材的质量评价,获得了可靠的评价结果。但是由于清热解毒口服液的上述特点和市场现状,其指纹图谱的构建依然有较多的困难和不确定性。
5.为解决上述问题,特提出本发明。
技术实现要素:
6.本发明的目的在于克服现有技术的不足,提供一种清热解毒口服液hplc指纹图谱和在清热解毒口服液质量控制中的应用。
7.本发明上述目的通过如下技术方案实现:
8.一种清热解毒口服液的hplc指纹图谱,基于如下色谱条件构建:
9.液相色谱仪为waterse2695高效液相色谱仪;
10.色谱柱为sapphirec18,规格为4.6mm*250mm、5μm;
11.流动相为甲醇-0.2%磷酸水溶液,梯度洗脱,流速1ml/min,柱温35℃,检测波长238nm,梯度洗脱程序如下,理论板数按黄芩苷峰计算应不得低于6000;
[0012][0013]
所述hplc指纹图谱的特征色谱峰为黄芩苷、绿原酸、3,5-二-o-咖啡酰奎宁酸、4,
5-二-o-咖啡酰奎宁酸、栀子苷、龙胆苦苷和连翘酯苷a峰,特征时段色谱图为5~100min色谱图。
[0014]
优选地,取清热解毒口服液适量,用70%甲醇稀释至5倍体积,摇匀,精密量取10μl注入液相色谱仪,测定,记录色谱图,构建清热解毒口服液的hplc指纹图谱。
[0015]
优选地,所述waterse2695高效液相色谱仪配备2998pda检测器。
[0016]
上述hplc指纹图谱在清热解毒口服液质量控制中的应用。
[0017]
优选地,所述的应用包括如下步骤:
[0018]
步骤s1,分别取若干份质量合格的清热解毒口服液进行hplc分析,通过其hplc指纹图谱建立清热解毒口服液的对照指纹图谱;其中,含有和不含有苯甲酸钠的清热解毒口服液的对照指纹图谱分别建立;
[0019]
步骤s2,取待检测清热解毒口服液进行hplc分析,将其hplc指纹图谱与所述对照指纹图谱比对,符合以下条件判定为合格:(1)待检测清热解毒口服液的hplc指纹图谱中呈现与对照指纹图谱中相应保留时间的特征色谱峰;(2)按中药色谱指纹图谱相似度评价系统计算5~100分钟的色谱峰,待检测清热解毒口服液的hplc指纹图谱与对照指纹图谱相似度不得低于0.90;其中,含有和不含有苯甲酸钠的待检测清热解毒口服液的hplc指纹图谱分别与含有和不含有苯甲酸钠的清热解毒口服液的对照指纹图谱进行比对。
[0020]
有益效果:
[0021]
本发明提供的清热解毒口服液的hplc指纹图谱可以有效用于控制清热解毒口服液的质量,可以克服传统质量控制方法难以全面、客观、准确、高效地评价其质量的缺陷。
附图说明
[0022]
图1为清热解毒口服液对照指纹图谱;图2为清热解毒口服液对照指纹图谱及混合对照图谱;图3和图4为全方对照、单味对照药材及其相应阴性样品图谱。
具体实施方式
[0023]
下面结合实施例具体介绍本发明实质性内容,但并不以此限定本发明的保护范围。
[0024]
实施例1:基于指纹图谱的质量控制
[0025]
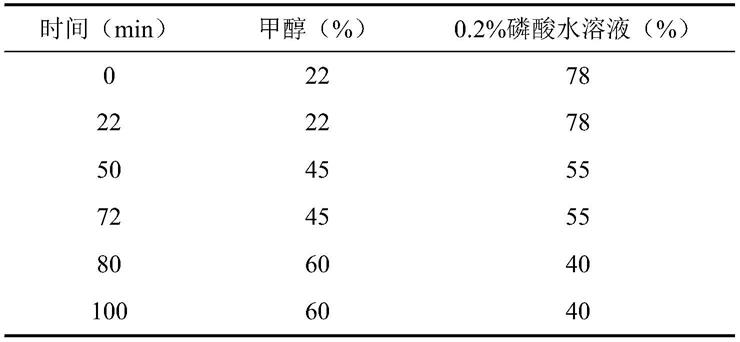
色谱条件waterse2695高效液相色谱仪(2998pda检测器),色谱柱:sapphirec18(4.6mm*250mm,5μm),流动相为甲醇(a)-0.2%磷酸水溶液(b),梯度洗脱,流速1ml/min,柱温35℃,进样量10μl,检测波长238nm,梯度洗脱程序详见表1流动相梯度洗脱表。理论板数按黄芩苷峰计算应不得低于6000。
[0026]
表1流动相梯度洗脱表
[0027][0028]
参照物溶液的制备取黄芩苷、绿原酸、3,5-二-o-咖啡酰奎宁酸、4,5-二-o-咖啡酰奎宁酸、栀子苷、龙胆苦苷、连翘酯苷a对照品适量,精密称定,加甲醇分别制成含黄芩苷170μg、绿原酸110μg、3,5-二-o-咖啡酰奎宁酸50μg、4,5-二-o-咖啡酰奎宁酸50μg、栀子苷110μg、龙胆苦苷100μg、连翘酯苷a100μg的混合对照溶液。
[0029]
供试品溶液的制备精密量取本品(即清热解毒口服液)1ml,置5ml量瓶中,加70%甲醇至刻度,摇匀,即得。
[0030]
测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,记录色谱图。供试品指纹图谱中,应分别呈现与参照物色谱保留时间相应的色谱峰。按中药色谱指纹图谱相似度评价系统计算5~100分钟的色谱峰,供试品指纹图谱与对照指纹图谱(图1)相似度不得低于0.90。图1中,s1为不含苯甲酸钠的清热解毒口服液的指纹图谱,s2为含苯甲酸钠的清热解毒口服液的指纹图谱,指纹图谱特征峰为:峰5
‑‑
绿原酸,峰7
‑‑
龙胆苦苷,峰9
‑‑
栀子苷,峰18
‑‑
连翘酯苷a,峰20
‑‑
3,5-二-o-咖啡酰奎宁酸,峰23
‑‑
4,5-二-o-咖啡酰奎宁酸,峰26
‑‑
黄芩苷。
[0031]
实施例2:指纹图谱构建过程
[0032]
1、仪器与试药
[0033]
仪器:waterse2695高效液相色谱仪(2998pda检测器)
[0034]
试药:甲醇和磷酸为色谱纯;水为纯化水;其它试剂均为分析纯。
[0035]
对照品:3,5-二-o-咖啡酰奎宁酸、黄芩苷、绿原酸、栀子苷、龙胆苦苷、哈巴俄苷、连翘苷、(r,s)-告依春、芒果苷、1,3-二咖啡酰奎宁酸,黄芩素、汉黄芩苷(批号:111782-201807、110715-201821、110753-202018、110749-201919、110770-201918、11730-201709,110821-201816,111753-201304,111607-201704,111717-201402,111595-201306,112002-201702,含量:94.3%、95.4%、96.1%、97.1%、97.1%、95.9%、95.1%、99.9%、98.1%、94.5%、97.8%、98.5%)购于中国食品药品检定研究院,含量测定用。4,5-二-o-咖啡酰奎宁酸、连翘酯苷a、异绿原酸b(批号:9113、9532、3089,含量:91.3%、98.5%、99.7%)购于上海施丹德标准技术服务有限公司。汉黄芩素(111514-201304)购于中国食品药品检定研究院,鉴别用。
[0036]
药材:黄芩、金银花、栀子、龙胆、连翘、地黄、板蓝根、知母、麦冬、甜地丁、石膏药材由部分抽样厂家寄送以及亳州药材市场收集。
[0037]
2、方法与结果
[0038]
2.1色谱条件
[0039]
2.1.1色谱柱的选择(耐用性考察)
[0040]
考察了如下色谱柱:
[0041]
①
sapphirec18(4.6mm*250mm,5μm);
[0042]
②
symmetryshieldrp18(4.6mm*250mm,5μm);
[0043]
③
zorbraxelipseplusc18(4.6mm*250mm,5μm);
[0044]
④
symmetryc18(4.6mm*250mm,5μm);
[0045]
⑤
sunfirec18(4.6mm*250mm,5μm);
[0046]
⑥
bostongreenods(4.6mm*250mm,5μm);
[0047]
⑦
diamonsilplusc18(4.6mm*250mm,5μm)。
[0048]
结合清热解毒口服液hplc含量测定中各组分分离情况,
①
、
②
、
⑤
、
⑦
色谱柱能将特征成分有效分离,且无干扰,但
①
色谱柱的其他各峰分离和分布情况更为理想,因此本方法建立以及样品检测采用
①
色谱柱。
[0049]
2.1.2流动相选择
[0050]
考察了乙腈-水、甲醇-水、乙腈-0.4%甲酸水溶液、甲醇-0.4%甲酸水溶液、甲醇-0.2%磷酸溶液、乙腈-0.2%磷酸水溶液等流动相系统,结果甲醇-0.2%磷酸溶液系统分离效果较好,优化梯度如表2。
[0051]
表2流动相梯度洗脱表
[0052][0053]
结果采用以上色谱柱及流动相,梯度洗脱时基线较平稳、色谱峰较多、峰型较好、分离效果良好。
[0054]
2.1.3检测波长的选择
[0055]
清热解毒口服液在指纹图谱中峰的贡献量(面积和数目):黄芩、金银花》连翘、栀子》龙胆、知母》地黄、板蓝根、麦冬、甜地丁》石膏,若以各药味特征成分最大吸收波长设置波长切换,黄芩苷峰太高,导致其他峰因响应或含量较低被淹没,参照清热解毒口服液hplc含量测定,选择238nm可规避这一问题,同时峰的数目较多,分布较为合理。
[0056]
2.2供试品溶液制备方法考察
[0057]
口服液处方中所有药味均经水提醇沉,提取出的有效成分极性较大,采用50%甲醇、70%甲醇、纯甲醇稀释结果差异不大。最终选择以70%甲醇稀释、定容。
[0058]
2.3指纹图谱建立
[0059]
因本次抽样口服液样品量前四位厂家在多组分含量分析中出现多个成分不合格,
且指纹图谱中出现多个峰丢失,为避免劣币驱逐良币,特结合薄层、多组分含量测定、地黄和板蓝根液质鉴别结果,选择上述项目皆符合规定且色谱峰信息丰富的厂家hplc指纹图谱,导入“中药色谱指纹图谱相似度评价系统”(2012.1版)进行色谱峰匹配,经处理后先建立各厂家对照指纹图谱,再建立清热解毒口服液对照指纹图谱。另外由于部分厂家加入苯甲酸钠作为防腐剂,该物质在238nm下有较强吸收,有无添加该防腐剂对相似度的计算影响很大,因此在建立对照指纹图谱时将两者分开。共标定34个共有峰,详见图2清热解毒口服液对照指纹图谱及混合对照图谱(其中,s1代表混合对照溶液,s2代表片剂,s3代表含苯甲酸钠口服液,s4代表不含苯甲酸钠口服液)。
[0060]
共有峰的指认:将清热解毒口服液处方中的12味药按处方中比例,模拟制备全方对照、单味对照药材及其相应阴性样品(除石膏),进样分析,通过与各药味部分特征成分对照品保留时间和光谱图进行比对,各共有峰的归属详见表3清热解毒口服液对照指纹图谱共有峰归属。地黄中地黄苷d和板蓝根中(r,s)-告依春因极性大,出峰时间早,溶剂峰或极性较大糖类对其造成较大干扰,因此采用液质联用进行鉴别。另外玄参、麦冬、甜地丁各有效成分在dad检测器下响应低,无法指认。结果色谱图详见图3-4全方对照、单味对照药材及其相应阴性样品图谱。
[0061]
表3清热解毒口服液对照指纹图谱共有峰归属
[0062][0063]
2.4方法学验证
[0064]
2.4.1精密度试验
[0065]
取吉林m(批号:20201001)样品,按正文下方法制备供试品溶液,以2.1项下色谱条件连续进样6次,记录7个标定共有峰面积和保留时间,以黄芩苷为参比峰,详见表4精密度相对峰面积考察结果,表5精密度相对保留时间考察结果,rsd均小于1%,精密度良好。
[0066]
表4精密度相对峰面积考察结果
[0067][0068]
表5精密度相对保留时间考察结果
[0069][0070]
2.4.2稳定性试验
[0071]
取吉林m(批号:20201001)样品,按正文下方法制备供试品溶液,分别于0、4、8、12、24、48h进样,色谱条件同2.1项下条件一致,记录7个标定共有峰面积和保留时间,以黄芩苷
为参比峰,详见表6稳定性相对峰面积考察结果,表7稳定性相对保留时间考察结果,rsd均小于1%,说明样品溶液在48h内稳定性良好。
[0072]
表6稳定性相对峰面积考察结果
[0073][0074]
表7稳定性相对保留时间考察结果
[0075][0076]
2.4.3重复性试验
[0077]
取吉林m(20201001)样品,共6份,进样条件不变,记录色谱图,以黄芩苷为参比峰,
计算各共有峰相对峰面积的rsd均小于3%,相对保留时间的rsd均小于1%,详见表8重复性相对峰面积考察结果、表9重复性相对保留时间考察结果,表明该方法重复性符合要求。
[0078]
表8重复性相对峰面积考察结果
[0079][0080]
表9重复性相对保留时间考察结果
[0081][0082]
2.5样品测定
[0083]
依法对200批样品进行测定,并与清热解毒口服液对照指纹图谱比较计算相似度。以0.90作为相似度合格判定限度,结果不含苯甲酸钠(111批)相似度为0.68~0.99,含苯甲酸钠(89批)为0.86~0.97。前者合格批数为66,合格率为59.5%,后者合格批数为81,合格
率为91.0%,口服液总合格率为73.5%。具体测定结果见表10清热解毒口服液指纹图谱项目检验结果汇总。
[0084]
表10清热解毒口服液指纹图谱项目检验结果汇总
[0085]
[0086]
[0087]
[0088][0089]
综上,本发明提供的清热解毒口服液的hplc指纹图谱可以有效用于控制清热解毒口服液的质量,可以克服传统质量控制方法难以全面、客观、准确、高效地评价其质量的缺陷。
[0090]
上述实施例的作用在于具体介绍本发明的实质性内容,但本领域技术人员应当知道,不应将本发明的保护范围局限于该具体实施例。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。