1.本发明涉及一种β-烟酰胺单核酸含量和有关物质的测定,属于药物分析技术领域。
背景技术:
2.β-烟酰胺单核苷酸(β-nicotimamide mononucletide,nmn)是人体内一种内源性物质,由烟酰胺在烟酰胺磷酸核糖转移酶的催化下生产,随后nmn又在烟酰胺单核苷酸转移酶的催化下生成烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,nad
)。nmn在人体内的生理作用都是通过体内转化为nad
实现,已有的可选研究表明,nmn主要包括参与多种疾病的预防,如2型糖尿病、肥胖症和心力衰竭等。2016年,代谢领域研究最有影响的刊物《cell metabolism》(mills等,2016,24:795
–
806)报道了给实验鼠补充饲喂nmn可有效减轻小鼠与年龄相关的生理衰竭,在没有任何明显毒性或有害影响的情况下,nmn可抑制与年龄相关的体重增加,可增加能量代谢,促进体力活动,改善胰岛素敏感性和血脂水平,并改善眼功能和其他病理性疾病,证明了nmn在抗衰老方面的巨大潜力。因此,nmn是医药、功能性食品、化妆品等行业的珍贵原料。
3.目前,nmn的制备方法主要包括以下三种:1、酵母菌发酵法;2、化学合成法;3、酶催化合成法。其中酶催化合成法是以烟酰胺、三磷酸腺苷和d-核糖作为底物,在核糖激酶(pk)和烟酰胺核糖磷酸转移酶(nampt)作为催化剂的作用下催化制备nmn,是一种绿色环保无公害的nmn制备方法。
4.在nmn的酶催化合成过程中会产生多个重要的有关物质,这些有关物质如果过多引入药物成品中,将可能导致严重的用药安全性问题。因此,在原料药合成阶段和制剂阶段都需要对这些有关物质进行严格控制。enrico balducci等人报道了用液相色谱法,色谱条件为十八烷基硅烷键合硅胶柱,100mmol/l、ph6.0的磷酸二氢钾缓冲盐和甲醇为流动相,柱温16.5℃,流速1.3ml/min,波长254nm,梯度洗脱方式对nmn进行分析(analytical biochemistry,1995,228,64-68)。leonardo sorci等人报道用100mmol/l磷酸二氢钾、8mmol/l四丁基溴化铵、ph6.0的缓冲盐和甲醇为流动相流速1.0ml/min,梯度洗脱方式对nmn进行分析(pnas,2009,106(9):3083-3088)。这两种方法流动相中缓冲盐浓度过大,易伤色谱柱,且特定杂质烟酰胺核糖(nr)与主峰nmn间不能基线分离,无法实现准确分析。因此寻求一种能够有效分离且尽可能多的检出nmn中有关物质的高效液相色谱法,对于nmn的精确质量控制具有十分重要的意义。
5.cn 113358776a公开了一种hplc-uv同时检测nmn中5种有关物质的方法,色谱柱采用c18-aq柱,流动相a为磷酸二氢钾缓冲盐溶液,流动相b为甲醇,洗脱方式为梯度洗脱,流速为0.5~1.5ml/min,柱温为15~35℃,可以定量检测nmn中三磷酸腺苷(atp)、二磷酸腺苷(adp)、腺苷酸(amp)、烟酰胺(na)和nho3的含量。烟酰胺核糖(nr)是合成nmn的起始物料,也是酸破坏和氧化降解nmn时易产生的降解产物,是nmn制备工艺中需要严格控制的特定杂质,但该专利未提及建立的方法是否可以用于同时检测nr。我们将该专利公开的方法用于
同时检测nmn、nr、na、atp、adp、amp和烟酸(vb3)时,发现nr和atp不能达到基线分离。因而,急需开发操作简便、专属性强、准确度高、适用性广、能同时检测nmn制备过程中多种有关物质的hplc方法,更好的控制nmn的品质,为食品安全提供保障。
技术实现要素:
6.本发明要解决的技术问题是提供一种能同时检测β-烟酰胺单核酸的含量和有关物质的方法。
7.为解决上述技术问题,本发明的β-烟酰胺单核酸含量和有关物质的测定方法包括:
8.采用hplc-uv法;所述hplc的色谱柱采用sinochrom ods-bp,4.6
×
250mm 5μm或柱效相当的色谱柱;流动相a为0.1~0.3%磷酸二氢钾和0.05~0.15%四丁基氢氧化铵的混合水溶液,流动相b为甲醇;流速为1
±
0.3ml/min,柱温为35℃
±
10℃;
9.所述β-烟酰胺单核酸含量的测定的洗脱梯度为等度或梯度,所述有关物质的测定的洗脱为梯度;
10.所述等度为流动相a运行7~10min;
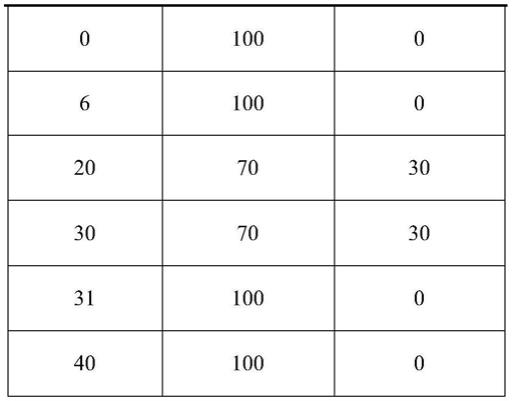
11.所述梯度为:0~6分钟,流动相a为100%,流动相b为0;6~20分钟,流动相a由100%变为70%,流动相b由0变为30%;20~30分钟,流动相a为70%,流动相b为30%;30~31分钟,流动相a由70%变为100%,流动相b由30%变为0;31~40分钟,流动相a为100%,流动相b为0;
12.所述uv检测波长为265
±
10nm。
13.在一种具体实施方式中,所述流动相a中磷酸二氢钾的浓度为0.2%。
14.在一种具体实施方式中,所述流动相a中四丁基氢氧化铵的浓度为0.1%。
15.在一种具体实施方式中,所述流速为1ml/min。
16.在一种具体实施方式中,所述柱温为35℃。
17.在一种具体实施方式中,所述检测波长为260nm。
18.在一种具体实施方式中,所述hplc的对照品和供试品溶液的制备:取nmn对照品适量和待测样品适量,分别用水溶解并定量稀释制成每1ml中约含50μg,作为对照品溶液和供试品溶液。
19.在一种具体实施方式中,所述的β-烟酰胺单核酸含量的测定方法,其特征在于,所述hplc的供试品溶液的制备:取待测的nmn样品,用水溶解并定量稀释制成每1ml中含0.5mg,作为供试品溶液;所述hplc的自身对照溶液的制备:取供试品溶液适量,加水溶解并定量稀释制成每1ml中含5μg,作为自身对照溶液。
20.在一种具体实施方式中,所述的β-烟酰胺单核酸有关物质的测定方法,其特征在于,所述有关物质为烟酰胺核糖(nr)、烟酰胺(na)、三磷酸腺苷(atp)、二磷酸腺苷(adp)、腺苷酸(amp)和烟酸(vb3)中的至少一种。
21.有益效果:
22.1.本发明溶剂和杂质均不干扰主峰的检出,各杂质之间均能达到基线分离,具体表现在可以定量检测nmn中烟酰胺核糖(nr)、烟酰胺(na)、三磷酸腺苷(atp)、二磷酸腺苷(adp)、腺苷酸(amp)和烟酸(vb3)等6个已知杂质的含量,其中杂质nr即是合成nmn的起始物
料,也是酸破坏、氧化易产生的降解杂质,是nmn合成工艺中需要严格控制的特定杂质,本发明能准确测定杂质nr的含量,方法操作简便,专属性强,准确度高,适用性广,可以更好的控制nmn的品质,为食品安全提供保障;
23.2.同时,本发明还有效避免了现有技术中流动相高盐浓度对色谱仪的损害。
附图说明
24.图1nmn有关物质测定——系统适用性溶液色谱图;
25.图2nmn有关物质测定——高温破坏样品溶液色谱图;
26.图3nmn有关物质测定——碱破坏样品溶液色谱图;
27.图4nmn有关物质测定——酸破坏样品溶液色谱图;
28.图5nmn有关物质测定——氧化破坏样品溶液色谱图;
29.图6nmn含量测定——系统适用性溶液色谱图;
30.图7nmn含量测定——线性关系图;
31.图8对比例1——系统适用性溶液色谱图
32.图9对比例2——系统适用性溶液色谱图
33.图10对比例3——系统适用性溶液色谱图
具体实施方式
34.为解决上述技术问题,本发明的β-烟酰胺单核酸含量和有关物质的测定方法包括:
35.采用hplc-uv法;所述hplc的色谱柱采用sinochrom ods-bp,4.6
×
250mm 5μm或柱效相当的色谱柱;流动相a为0.1~0.3%磷酸二氢钾和0.05~0.15%四丁基氢氧化铵的混合水溶液,流动相b为甲醇;流速为1
±
0.3ml/min,柱温为35℃
±
10℃;
36.所述β-烟酰胺单核酸含量的测定的洗脱梯度为等度或梯度,所述有关物质的测定的洗脱为梯度;
37.所述等度为流动相a运行7~10min;
38.所述梯度为:0~6分钟,流动相a为100%,流动相b为0;6~20分钟,流动相a由100%变为70%,流动相b由0变为30%;20~30分钟,流动相a为70%,流动相b为30%;30~31分钟,流动相a由70%变为100%,流动相b由30%变为0;31~40分钟,流动相a为100%,流动相b为0;
39.所述uv检测波长为265
±
10nm。
40.在一种具体实施方式中,所述流动相a中磷酸二氢钾的浓度为0.2%。
41.在一种具体实施方式中,所述流动相a中四丁基氢氧化铵的浓度为0.1%。
42.在一种具体实施方式中,所述流速为1ml/min。
43.在一种具体实施方式中,所述柱温为35℃。
44.在一种具体实施方式中,所述检测波长为260nm。
45.在一种具体实施方式中,所述hplc的对照品和供试品溶液的制备:取nmn对照品适量和待测样品适量,分别用水溶解并定量稀释制成每1ml中约含50μg,作为对照品溶液和供试品溶液。
46.在一种具体实施方式中,所述的β-烟酰胺单核酸含量的测定方法,其特征在于,所述hplc的供试品溶液的制备:取待测的nmn样品,用水溶解并定量稀释制成每1ml中含0.5mg,作为供试品溶液;所述hplc的自身对照溶液的制备:取供试品溶液适量,加水溶解并定量稀释制成每1ml中含5μg,作为自身对照溶液。
47.在一种具体实施方式中,所述的β-烟酰胺单核酸有关物质的测定方法,其特征在于,所述有关物质为烟酰胺核糖(nr)、烟酰胺(na)、三磷酸腺苷(atp)、二磷酸腺苷(adp)、腺苷酸(amp)和烟酸(vb3)中的至少一种。
48.下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实施例范围之中。
49.实施例1
50.(1)供试品溶液的制备:取nmn适量,精密称定,用水溶解并定量稀释制成每1ml中约含0.5mg的溶液,作为供试品溶液;
51.(2)自身对照溶液的制备:精密量取供试品溶液适量,加水定量稀释制成每1ml中约含5μg的溶液,作为自身对照溶液;
52.(3)检测:分别取供试品溶液、自身对照溶液注入高效液相色谱仪,其中色谱条件如下:
53.色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相a为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,流动相b为甲醇;流速为1ml/min,柱温35℃,检测波长为260nm。洗脱梯度为:
54.表1:实施例1洗脱梯度
55.时间(分钟)流动相a(%)流动相b(%)0100061000207030307030311000401000
56.实施例2
57.(1)供试品溶液的制备:取nmn适量,精密称定,用水溶解并定量稀释制成每1ml中约含50μg的溶液,作为供试品溶液;
58.(2)对照品溶液的制备:精密量取nmn对照品适量,加水定量稀释制成每1ml中约含50μg的溶液,作为对照品溶液;
59.(3)检测:分别取供试品溶液、对照品溶液注入高效液相色谱仪,其中色谱条件如下:
60.色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,等度洗脱;流速为1ml/min,柱温35℃,检测波长为265nm。
61.实施例3
62.样品制备同实施例1。
63.色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相a为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,流动相b为甲醇;流速为1ml/min,柱温25℃,检测波长为260nm。洗脱梯度为:
64.表2:实施例3洗脱梯度
[0065][0066][0067]
实施例4
[0068]
样品制备同实施例1。
[0069]
色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相a为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,流动相b为甲醇;流速为1ml/min,柱温45℃,检测波长为260nm。洗脱梯度为:
[0070]
表3:实施例4洗脱梯度
[0071]
时间(分钟)流动相a(%)流动相b(%)0100061000207030307030311000401000
[0072]
实施例5
[0073]
样品制备同实施例1。
[0074]
色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相a为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,流动相b为甲醇;流速为0.7ml/min,柱温35℃,检测波长为260nm。洗脱梯度为:
[0075]
表4:实施例5洗脱梯度
[0076]
[0077][0078]
实施例6
[0079]
样品制备同实施例1。
[0080]
色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相a为0.2%磷酸二氢钾和0.1%四丁基氢氧化铵的混合溶液,流动相b为甲醇;流速为1.3ml/min,柱温35℃,检测波长为260nm。洗脱梯度为:
[0081]
表5:实施例6洗脱梯度
[0082]
时间(分钟)流动相a(%)流动相b(%)0100061000207030307030311000401000
[0083]
实施例7
[0084]
色谱条件同实施例1。
[0085]
实验步骤:
[0086]
1.专属性试验
[0087]
取nr、na、amp、adp、atp、烟酸和nmn适量,用水稀释制成含各50μg的混合溶液,过滤后取续滤液作为供试品溶液,取供试品溶液20μl按照实施例1中的色谱条件进行色谱检测,图谱见图1。
[0088]
表6:实施例7的nmn与六个工艺杂质保留时间与分离度
[0089]
样品保留时间(min)分离度nr3.682/nmn4.9606.2na13.59041.8amp20.75134.4adp22.7268.7
atp29.57025.9烟酸36.0579.0
[0090]
通过计算分析可知,nmn与nr出峰最接近,分离度大于5.0,因此nmn与nr完全分离。nmn的理论塔板数为8304,大于5000,其他峰均不干扰nmn的检出,且各杂质峰均能被检出。该有关物质方法可用于检测这六个工艺杂质的检出。
[0091]
2.强制降解试验
[0092]
(1)高温破坏样品检测:精密称取nmn 12.5mg至25ml量瓶,加水溶解并稀释至刻度,60℃加热4小时,过滤后取续滤液作为供试品溶液,取供试品溶液20μl按照实施例1中的色谱条件进行色谱检测,图谱见图2。
[0093]
(2)碱破坏样品检查:精密称取nmn 12.5mg至25ml量瓶,加0.2mol/l氢氧化钠1ml,室温放置10min,加0.2mol/l盐酸1ml,用水稀释至刻度,过滤后取续滤液作为供试品溶液,取供试品溶液20μl按照实施例1中的色谱条件进行色谱检测,图谱见图3。
[0094]
(3)酸破坏样品检查:精密称取nmn12.5mg至25ml量瓶,加0.2mol/l盐酸1ml,室温放置30min,加0.2mol/l氢氧化钠1ml,用水稀释至刻度,过滤后取续滤液作为供试品溶液,取供试品溶液20μl按照实施例1中的色谱条件进行色谱检测,图谱见图4。
[0095]
(4)氧化破坏样品检查:精密称取nmn 12.5mg至25ml量瓶,加5%双氧水1ml,室温放置30min,用水稀释至刻度,过滤后取续滤液作为供试品溶液,取供试品溶液20μl按照实施例1中的色谱条件进行色谱检测,图谱见图5。
[0096]
由强制降解试验得到的色谱图可以发现,nmn在酸、氧化降解条件下均降解出了特定杂质nr,高温破坏、碱破坏条件下均有较大程度的降解。各个降解杂质与主峰之间均得到很好的分离,本发明有关物质分析方法可用于检测降解杂质。
[0097]
实施例8
[0098]
色谱条件同实施例2。
[0099]
实验步骤:
[0100]
(1)系统适应性溶液的制备:取nr和nmn适量,用水稀释制成含各杂质50μg的混合溶液作为供试品溶液,取供试品溶液20μl按照实施例2中的色谱条件进行色谱检测,图谱见图6。
[0101]
(2)线性溶液的制备:取nmn对照品适量,用水溶解并稀释制成0.2mg/ml,作线性储备液,将此溶液用水稀释制成浓度为66.6、50.0、40.0、23.5、18.2、9.5、6.4μg/ml的线性溶液,取各溶液20μl按照实施例2中的色谱条件进行色谱检测,线性关系图见图7。
[0102]
(3)精密度试验溶液:取nmn线性溶液浓度为50μg/ml的溶液20μl,按照实施例2中的色谱条件进行色谱检测。
[0103]
(4)回收率试验溶液的制备:取nmn对照品适量,加水溶解并稀释制成浓度为50μg/ml的溶液,作对照品溶液。取nmn样品适量,用水稀释制成浓度分别为60μg/ml、50μg/ml、40μg/ml的溶液,每个浓度各配置三份。取各试验溶液20μl按照实施例2中的色谱条件进行色谱检测。
[0104]
由系统适用性溶液可知,nr与nmn的分离度为15.37,分离良好,nmn理论塔板数为6107.3,大于5000;由线性试验可知,nmn浓度范围在6.4~200μg/ml范围内,r2为0.9997,线性关系良好;由精密度试验可知,试验溶液连续测定6次,峰面积rsd为0.19%,精密度良好;
由回收率试验可知,在80%、100%、120%三个浓度下,nmn的回收率在99.1.3%~101.9%之间,rsd%为2.73%,证实方法有良好准确度。综上所述,该方法可以准确可靠的测定nmn的含量。
[0105]
对比例1
[0106]
根据enrico balducci等人报道的hplc方法进行操作(analytical biochemistry,1995,228:64-68)。
[0107]
系统适用性溶液的制备:取nr和nmn适量,用水稀释制成含nmn 200μg和含nr 50μg的混合溶液。
[0108]
取系统适用性溶液10μl注入高效液相色谱仪,其中色谱条件如下:
[0109]
色谱柱为sinochrom ods-bp,4.6
×
250mm 5μm;流动相为0.1mol/l磷酸二氢钾,用磷酸或氢氧化钠调ph为6.0-甲醇(95:5);流速为1.3ml/min,柱温20℃,检测波长为260nm。洗脱梯度,色谱图见图8。
[0110]
对比例2
[0111]
根据专利cn 113358776 a中报道的hplc方法进行操作。
[0112]
取nr、na、amp、adp、atp、烟酸和nmn适量,用水稀释制成含各50μg的混合溶液,过滤后取续滤液作为系统适用性溶液。
[0113]
取系统适用性溶液20μl注入高效液相色谱仪,其中色谱条件如下:
[0114]
色谱柱为welchultimate aq-c18,4.6
×
250mm 5μm;流动相a为0.05mol/l磷酸二氢钾,用磷酸或氢氧化钠调ph为4.5,流动相b为甲醇;流速为0.5ml/min,柱温25℃,检测波长为260nm。洗脱梯度,梯度如下,色谱图见图9。
[0115]
表7:对比例2洗脱梯度
[0116]
时间(分钟)流动相a(%)流动相b(%)0100010100020505025208030208030.11000351000
[0117]
对比例3
[0118]
根据leonardo sorci等人报道的hplc方法进行操作(pnas,2009,106(9):3083-3088)。
[0119]
系统适用性溶液的制备:取nr和nmn适量,用水稀释制成含nmn 200μg和含nr 50μg的混合溶液。
[0120]
取系统适用性溶液10μl注入高效液相色谱仪,其中色谱条件如下:
[0121]
色谱柱为kromasil c18,4.6
×
250mm 5μm;流动相为0.1mol/l磷酸二氢钾和8mmol/l四丁基溴化铵的混合水溶液,用磷酸或氢氧化钠调ph为6.0-甲醇(95:5);流速为1ml/min,柱温30℃,检测波长为260nm。洗脱梯度,色谱图见图10。
[0122]
试验结果:
[0123]
实施例1、3、4、5、6考察不同流速、不同柱温对nmn与各杂质之间的分离度影响,结果表明,流速为0.7~1.3ml/min、柱温为25~45℃范围内变化,各杂质之间、杂质与nmn之前的分离度均大于2.0,说明方法耐用性良好。实施例7,nmn在有关物质检查方法下的强制降解试验中,nmn在酸、氧化降解条件下均降解出了特定杂质nr,高温破坏、碱破坏条件下均有较大程度的降解。各个降解杂质与主峰之间均得到很好的分离,说明方法专属性良好,能准确测定nmn中有关物质的含量。
[0124]
实施例2,在nmn有关物质控制方法上,将梯度改为等度,运行时间10min,大大节约nmn含量测定时间。实施例8,nmn含量测定方法经过验证,nmn浓度范围在6.4~200μg/ml范围内,r2为0.9997,线性关系良好;试验溶液连续测定6次,峰面积rsd为0.19%,精密度良好;在80%、100%、120%三个浓度下,nmn的回收率在99.1.3%~101.9%之间,rsd%为2.73%,证实方法有良好准确度。综上所述,该方法可以准确可靠的测定nmn的含量。
[0125]
对比例1中,nr与nmn不能基线分离,且出峰过快,溶剂易干扰检测;对比例2中,hplc流速降为0.5ml/min时,杂质nr与atp均不能基线分离,因此不能准确测定nr与atp的残留量;对比例3中,nr与nmn仍不能基线分离。
[0126]
由于nr是nmn合成工艺的起始物料,易残留在原料中,且原料nmn在氧化、酸破坏下易产生此杂质,因此nr是nmn原料中最重要的工艺和降解杂质,需要严格控制其残留量。atp是nmn酶法合成过程中易产生的工艺杂质,也要严格控制其残留量。采用本发明检测方法,各杂质之间分离度符合要求,各杂质也不干扰nmn的检出、特别是杂质nr不干扰nmn的检测,nr与atp之间分离度良好。
[0127]
综上所述,本发明显优于enrico balducci等人报道的液相色谱方法(对比例1)、优于cn 113358776 a专利中一种hplc-uv同时检测nmn中多种有关物质的方法(对比例2)、优于leonardo sorci等人报道的液相色谱方法(对比例3),选择本发明的检测方法可以更准确可靠的控制nmn的质量。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。