一种瑞舒伐他汀钙晶型a的制备方法
技术领域
1.本发明涉及药物化学领域,具体涉及一种瑞舒伐他汀钙晶型a的制备方法。
背景技术:
2.瑞舒伐他汀钙为他汀类调脂药,是hmg-coa还原酶抑制剂(羟甲基戊二酰辅酶a还原酶抑制剂,h ydroxy methylglutaryl coenzyme a reductase inhibitor),其结构与hmg-coa具有相似的结构片段,能竞 争性地抑制hmg-coa还原酶,hmg-coa还原酶是胆固醇合成过程中的限速酶,抑制hmg-coa还原酶, 导致肝细胞内胆固醇含量降低,反馈性刺激肝细胞表面低密度脂蛋白(ldl)受体表达增加,促进ldl 和ldl前体从循环中清除。
3.瑞舒伐他汀钙存在多种结晶形式,其中wo2000042024公开的晶型a的制备工艺为:以(e)-(6-{2-[4-(4
ꢀ‑
氟苯基)-6-异丙基-2-[甲基(甲磺酰基)氨基]嘧啶-5-基]乙烯基}(4r,6s)-2,2-二甲基[1,3]二恶烷-4-基)乙酸的(1 ~6c)烷基酯作为原料,在水溶性有机溶剂中进行酸水解和碱水解,水解完毕后,用非水溶性有机溶剂萃取, 得到瑞舒伐他汀钠盐水溶液,需要减压蒸馏去除有机溶剂后滴加钙盐,得到无定形瑞舒伐他汀钙;再用无 定形瑞舒伐他汀钙加入到水和有机溶剂的混合溶液中,加热搅拌,通过转晶后得到晶形a产品。该方法需 要通过复杂繁琐的后处理操作才能得到无定形瑞舒伐他汀钙,再用无定形瑞舒伐他汀钙进行转晶后才得到 瑞舒伐他汀钙晶型a,增加了操作步骤,不利于商业化生产。
[0004]
本发明研究一种瑞舒伐他汀钙晶型a的制备方法,所述方法中,中间体无需分离纯化,可仅使用两种 绿色环保试剂,一次分离操作,高收率(92%以上)、高纯度(99%以上)制得晶型a产品,所述方法避 免了多步反应先制得无定型再转晶成晶型a的多步繁琐操作。本发明提供的方法操作简便,生产周期短, 绿色环保,使成本大幅降低,适合商业化生产。
技术实现要素:
[0005]
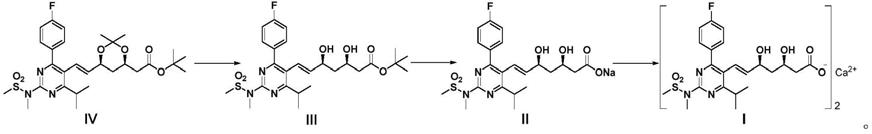
本发明提供一种瑞舒伐他汀钙a晶型的制备方法,包括以下步骤:
[0006]
1)化合物iv在非水溶液中与酸进行水解反应得到化合物iii;
[0007]
2)化合物iii与碱如氢氧化钠进行水解反应得到化合物ii;和
[0008]
3)化合物ii在水与有机溶剂的混合溶剂中与水溶性钙盐反应,制备得到瑞舒伐他汀钙晶型a;
[0009]
反应路线如下:
[0010][0011]
所述方法中,可以分离中间体化合物iii和/或化合物ii;也可不分离中间体化合物iii和/或化合物ii,优选 不分离中间体化合物iii和/或化合物ii。
[0012]
步骤1)中,所述非水溶液包括选自甲基叔丁基醚、乙醚、异丙醚、四氢呋喃、2-甲基
四氢呋喃中的 至少一种。在一些实施例中,所述非水溶液优选为甲基叔丁基醚。
[0013]
步骤1)中,所述酸可为任意适宜浓度的无机酸。在一些实施方式中,所述酸包括选自盐酸或者硫酸 中的至少一种。在一些实施方式中,所述酸包括选自浓盐酸或者浓硫酸中的至少一种。在一些实施例中, 所述酸为浓盐酸。在一些实施例中,所述酸为浓硫酸。
[0014]
步骤1)中,所述化合物iv与酸的投料质量比为1.0:0.1~1.0。在一些实施例中,所述化合物iv与 酸的投料质量比为1.0:0.2~1.0;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.3~1.0; 在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.4~1.0;在一些实施例中,所述化合物iv与 酸的投料质量比为1.0:0.5~1.0;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.6~1.0; 在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.7~1.0;在一些实施例中,所述化合物iv与 酸的投料质量比为1.0:0.8~1.0;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.9~1.0。 在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.9~1.0。在一些实施例中,所述化合物iv与 酸的投料质量比为1.0:0.1;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.2;在一些实施 例中,所述化合物iv与酸的投料质量比为1.0:0.3;在一些实施例中,所述化合物iv与酸的投料质量比 为1.0:0.4;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.5;在一些实施例中,所述化合 物iv与酸的投料质量比为1.0:0.6;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.7;在 一些实施例中,所述化合物iv与酸的投料质量比为1.0:0.8;在一些实施例中,所述化合物iv与酸的投 料质量比为1.0:0.9;在一些实施例中,所述化合物iv与酸的投料质量比为1.0:1.0。
[0015]
步骤1)中,所述化合物iv与非水溶液的投料质量比为1.0:3.0~20.0。在一些实施例中,所述化合 物iv与非水溶液的投料质量比为1.0:3.0~18.0;在一些实施例中,所述化合物iv与非水溶液的投料质 量比为1.0:3.0~15.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:3.0~10.0;在 一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:3.0~5.0;在一些实施例中,所述化合物 iv与非水溶液的投料质量比为1.0:5.0~20.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比 为1.0:5.0~15.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:5.0~10.0;在一些 实施例中,所述化合物iv与非水溶液的投料质量比为1.0:5.0~8.0;在一些实施例中,所述化合物iv与 非水溶液的投料质量比为1.0:10.0~20.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为 1.0:15.0~20.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:3.0;在一些实施例中, 所述化合物iv与非水溶液的投料质量比为1.0:5.0;在一些实施例中,所述化合物iv与非水溶液的投料 质量比为1.0:8.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:10.0;在一些实施 例中,所述化合物iv与非水溶液的投料质量比为1.0:12.0;在一些实施例中,所述化合物iv与非水溶液 的投料质量比为1.0:15.0;在一些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:18.0;在一 些实施例中,所述化合物iv与非水溶液的投料质量比为1.0:20.0。
[0016]
步骤1)中,所述化合物iv与甲基叔丁基醚的投料质量比为1.0:5.0~15.0。在一些实施例中,所述 化合物iv与甲基叔丁基醚的投料质量比为1.0:5.0~13.0;在一些实施例中,所述化合物iv与甲基叔丁 基醚的投料质量比为1.0:5.0~11.0;在一些实施例中,所述化合物iv与甲基叔丁基醚的投料质量比为1.0: 5.0~9.0;在一些实施例中,所述化合物iv
与甲基叔丁基醚的投料质量比为1.0:5.0~7.0;在一些实施例 中,所述化合物iv与甲基叔丁基醚的投料质量比为1.0:5.0;在一些实施例中,所述化合物iv与甲基叔 丁基醚的投料质量比为1.0:6.0;在一些实施例中,所述化合物iv与甲基叔丁基醚的投料质量比为1.0: 8.0;在一些实施例中,所述化合物iv与甲基叔丁基醚的投料质量比为1.0:10.0;在一些实施例中,所述 化合物iv与甲基叔丁基醚的投料质量比为1.0:12.0;在一些实施例中,所述化合物iv与甲基叔丁基醚的 投料质量比为1.0:15.0。
[0017]
本发明涉及的合成化合物iii的反应,可以采用高效液相色谱法(hplc)监控反应终点,当检测表明 反应原料化合物iv少于其1.0%时,视为反应结束,反应时间通常在12h以下。
[0018]
步骤2)中,所述碱包括选自氢氧化钠或者氢氧化钾中至少一种。所述碱可采用水溶液的形式加入反 应体系中,所述碱的水溶液可为任意适宜的浓度。在一些实施例中,所述碱为氢氧化钾。在一些实施例中, 所述碱优选为氢氧化钠。
[0019]
步骤2)中,所述化合物iv与碱的投料质量比可以为1.0:0.07~1.0。在一些实施例中,所述化合物 iv与碱的投料质量比为1.0:0.1~1.0。在一些实施例中,所述化合物iv与碱的投料质量比为1.0:0.1~ 0.8;在一些实施例中,所述化合物iv与碱的投料质量比为1.0:0.1~0.6;在一些实施例中,所述化合物 iv与碱的投料质量比为1.0:0.1~0.4;在一些实施例中,所述化合物iv与碱的投料质量比为1.0:0.1~ 0.2;在一些实施例中,所述化合物iv与碱的投料质量比为1.0:0.1;在一些实施例中,所述化合物iv与 碱的投料质量比为1.0:0.3;在一些实施例中,所述化合物iv与碱的投料质量比为1.0:0.5;在一些实施 例中,所述化合物iv与碱的投料质量比为1.0:0.7;在一些实施例中,所述化合物iv与碱的投料质量比 为1.0:0.9;在一些实施例中,所述化合物iv与碱的投料质量比为1.0:1.0。
[0020]
步骤2)中,反应结束后,将反应液静置分层,取水层用冰乙酸调节ph=8~10,直接用于步骤3)。
[0021]
在一些实施例中,所述水层ph用冰乙酸调节为8。在一些实施例中,所述水层ph用冰乙酸调节为8.5。
[0022]
在一些实施例中,所述水层ph用冰乙酸调节为9。在一些实施例中,所述水层ph用冰乙酸调节为9.5。
[0023]
在一些实施例中,所述水层ph用冰乙酸调节为10。
[0024]
本发明涉及的合成化合物ii的反应,可以采用高效液相色谱法(hplc)监控反应终点,当检测表明 反应原料化合物iii少于其0.5%时,视为反应结束,反应时间通常在5h以下。
[0025]
步骤3)中,所述水溶性钙盐包括选自氯化钙、乙酸钙中的至少一种。在一些实施例中,所述水溶性 钙盐为氯化钙。在一些实施例中,所述水溶性钙盐为乙酸钙。
[0026]
基于化合物iv的投料量计算,步骤3)中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.09~ 1.0。在一些实施例中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.1~0.8;在一些实施例中,所 述化合物iv与水溶性钙盐的投料质量比为1.0:0.1~0.6;在一些实施例中,所述化合物iv与水溶性钙盐 的投料质量比为1.0:0.1~0.4;在一些实施例中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.1~ 0.2;在一些实施例中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.1;在一些实施例中,所述化 合物iv与水溶
性钙盐的投料质量比为1.0:0.3;在一些实施例中,所述化合物iv与水溶性钙盐的投料质 量比为1.0:0.1;在一些实施例中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.5;在一些实施例 中,所述化合物iv与水溶性钙盐的投料质量比为1.0:0.7;在一些实施例中,所述化合物iv与水溶性钙 盐的投料质量比为1.0:0.9;在一些实施例中,所述化合物iv与水溶性钙盐的投料质量比为1.0:1.0。
[0027]
步骤3)中,所述化合物ii与水溶性钙盐反应的反应溶剂为水与有机溶剂的混合溶剂,所述有机溶剂 包括选自甲醇,乙醇,异丙醇和叔丁醇中的至少一种。
[0028]
步骤3)中,所述化合物iv与有机溶剂的投料质量比为1.0:1.5~10.0。在一些实施例中,所述化合 物iv与有机溶剂的投料质量比为1.0:2.0~8.0。在一些实施例中,所述化合物iv与有机溶剂的投料质量 比为1.0:2.0~6.0。在一些实施例中,所述化合物iv与有机溶剂的投料质量比为1.0:2.0~4.0。在一些 实施例中,所述化合物iv与有机溶剂的投料质量比为1.0:2.0;在一些实施例中,所述化合物iv与有机 溶剂的投料质量比为1.0:3.0;在一些实施例中,所述化合物iv与有机溶剂的投料质量比为1.0:5.0;在 一些实施例中,所述化合物iv与有机溶剂的投料质量比为1.0:7.0;在一些实施例中,所述化合物iv与 有机溶剂的投料质量比为1.0:9.0;在一些实施例中,所述化合物iv与有机溶剂的投料质量比为1.0:10.0。
[0029]
步骤3)中,化合物ii与水溶性钙盐反应中,化合物iv与水的投料质量比为1.0:8.0~15.0。在一些 实施例中,所述化合物iv与水的投料质量比为1.0:8.0~13.0;在一些实施例中,所述化合物iv与水的 投料质量比为1.0:8.0~11.0;在一些实施例中,所述化合物iv与水的投料质量比为1.0:8.0~9.0。在一 些实施例中,所述化合物iv与水的投料质量比为1.0:8.0;在一些实施例中,所述化合物iv与水的投料 质量比为1.0:10.0;在一些实施例中,所述化合物iv与水的投料质量比为1.0:12.0;在一些实施例中, 所述化合物iv与水的投料质量比为1.0:13.0。
[0030]
步骤3)中,化合物ii与水溶性钙盐反应,在反应结束后,将反应液保温养晶0.5h-4h,然后降温至 10℃-35℃析晶,过滤,滤饼干燥,得到瑞舒伐他汀钙晶型a。在一些实施方式中,化合物ii与水溶性钙 盐反应,在反应结束后,将反应液保温养晶2h,然后降温至30℃析晶,过滤,滤饼干燥,得到瑞舒伐他 汀钙晶型a。
[0031]
本发明所述方法中,各步骤反应的反应温度范围分别为20℃~90℃。在一些实施例中,所述反应温度 为20℃~80℃;在一些实施例中,所述反应温度为20℃~70℃;在一些实施例中,所述反应温度为20℃~ 60℃;在一些实施例中,所述反应温度为20℃~50℃;在一些实施例中,所述反应温度为20℃~40℃; 在一些实施例中,所述反应温度为20℃~30℃;在一些实施例中,所述反应温度为30℃~90℃;在一些 实施例中,所述反应温度为40℃~90℃;在一些实施例中,所述反应温度为50℃~90℃;在一些实施例 中,所述反应温度为60℃~90℃;在一些实施例中,所述反应温度为70℃~90℃;在一些实施例中,所 述反应温度为80℃~90℃。在一些实施例中,所述反应温度为20℃;在一些实施例中,所述反应温度为 30℃;在一些实施例中,所述反应温度为40℃;在一些实施例中,所述反应温度为50℃;在一些实施例 中,所述反应温度为60℃;在一些实施例中,所述反应温度为70℃;在一些实施例中,所述反应温度为 80℃;在一些实施例中,所述反应温度为90℃。
[0032]
本发明所提供的制备方法,中间体无需分离纯化,获得的产物纯度高、收率高;反
应时间短,能耗低, 操作简单,可以降低成本,有利于工业化实施。
[0033]
术语定义
[0034]
在本发明的描述中,需要理解的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示 相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐 含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具 体的限定。
[0035]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例
”ꢀ
等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例 或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的 具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互 矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特 征进行结合和组合。
[0036]
本发明中,如“化合物i”和“式i所示的化合物”的表述,表示的是同一个化合物。
[0037]
x射线粉末衍射(xrpd)可检测晶型的变化、结晶度、晶构状态等信息,是鉴别晶型的常用手段。 xrpd图谱的峰位置主要取决于晶型的结构,对实验细节相对不敏感,而其相对峰高取决于与样品制备和 仪器几何形状有关的许多因素。因此,在一些实施方案中,本发明的晶型的特征在于具有某些峰位置的 xrpd图,同时,xrpd图谱的2θ的量度可以有实验误差,不同仪器以及不同样品之间,xrpd图谱的2θ 的量度可能会略有差别,因此所述2θ的数值不能视为绝对的。根据本试验所用仪器状况,衍射峰存在
±
0.2
°ꢀ
的误差容限。
具体实施方式
[0038]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例,对本发 明作进一步的详细说明。
[0039]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0040]
本发明中,℃表示摄氏度,h表示小时,g表示克,ml表示毫升,mtbe表示甲基叔丁基醚。
[0041]
实施例1
[0042][0043]
合成化合物iii
[0044]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h 后取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0045]
合成化合物ii
[0046]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反
应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0047]
合成化合物i
[0048]
向化合物ii水液加入400ml异丙醇,搅拌升温至55℃,滴加入110g10%氯化钙水溶液,保温养晶 2h,降温至30℃析晶,过滤,滤饼干燥后得到81.64g白色固体化合物i,纯度99.32%,收率94.22%。
[0049]
实施例2
[0050][0051]
合成化合物iii
[0052]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0053]
合成化合物ii
[0054]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0055]
合成化合物i
[0056]
向化合物ii水液加入400ml乙醇,搅拌升温至40℃,滴加入110g10%氯化钙水溶液,保温养晶2h, 降温至25℃析晶,过滤,滤饼干燥后得到81.40g白色固体化合物i,纯度99.56%,收率93.94%。
[0057]
实施例3
[0058][0059]
合成化合物iii
[0060]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0061]
合成化合物ii
[0062]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0063]
合成化合物i
[0064]
向化合物ii水液加入200ml甲醇,搅拌升温至35℃,滴加入110g10%氯化钙水溶液,保温养晶2h, 降温至30℃析晶,过滤,滤饼干燥后得到81.15g白色固体化合物i,纯度
99.44%,收率93.66%。
[0065]
实施例4
[0066][0067]
合成化合物iii:
[0068]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0069]
合成化合物ii
[0070]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0071]
合成化合物i
[0072]
向化合物ii水液加入700ml叔丁醇,搅拌升温至80℃,滴加入110g10%氯化钙水溶液,保温养晶2h, 降温至30℃析晶,过滤,滤饼干燥后得到78.09g白色固体化合物i,纯度99.21%,收率90.12%。
[0073]
实施例5
[0074][0075]
合成化合物iii
[0076]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0077]
合成化合物ii
[0078]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0079]
合成化合物i
[0080]
向化合物ii水液加入500ml异丙醇,搅拌升温至45℃,滴加入160g10%乙酸钙水溶液,保温养晶2h, 降温至25℃析晶,过滤,滤饼干燥后得到81.28g白色固体化合物i,纯度99.36%,收率93.81%。
[0081]
实施例6
[0082][0083]
合成化合物iii
[0084]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0085]
合成化合物ii
[0086]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0087]
合成化合物i
[0088]
向化合物ii水液加入300ml乙醇,搅拌升温至50℃,滴加入160g10%乙酸钙水溶液,保温养晶2h, 降温至15℃析晶,过滤,滤饼干燥后得到81.59g白色固体化合物i,纯度99.58%,收率94.16%。
[0089]
实施例7
[0090][0091]
合成化合物iii
[0092]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0093]
合成化合物ii
[0094]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0095]
合成化合物i
[0096]
向化合物ii水液加入400ml甲醇,搅拌升温至35℃,滴加入160g10%乙酸钙水溶液,保温养晶2h, 降温至20℃析晶,过滤,滤饼干燥后得到81.15g白色固体化合物i,纯度99.44%,收率93.66%。
[0097]
实施例8
[0098][0099]
合成化合物iii
[0100]
2l四口瓶中加入700ml甲基叔丁基醚、100g化合物iv和18.20g浓盐酸,控温30℃搅拌反应12h后 取样测hplc,反应至化合物iv≤1.0%,反应结束,得到化合物iii反应液,直接用于下一步;
[0101]
合成化合物ii
[0102]
向化合物iii反应液加入900g 2%naoh水溶液,控温30℃反应5h后取样测hplc,反应至化合物 iii≤0.5%,反应结束,静置分层,水层用冰乙酸调节ph=9,得到化合物ii水液,直接用于下一步;
[0103]
合成化合物i
[0104]
向化合物ii水液加入600ml叔丁醇,搅拌升温至60℃,滴加入160g10%乙酸钙水溶液,保温养晶2h, 降温至30℃析晶,过滤,滤饼干燥后得到80.09g白色固体化合物i,纯度99.03%,收率92.43%。
[0105]
实施例9晶型鉴定
[0106]
对上述实施例制备得到的化合物i进行x射线粉末衍射(xrpd)检测,并且和中国专利cn99815504.7 中所述的晶型a进行比对,相应的检测结果如下表所示:
[0107]
化合物i:
[0108][0109]
专利cn99815504.7晶型a:
[0110][0111]
根据上述xrpd检测结果可知,本发明方法所制备的化合物i为晶型a。
[0112]
仪器参数,测试条件及表征结果
[0113]
仪器信息:
[0114]
x射线粉末衍射分析仪
‑‑
panalytical
[0115]
测试方法:
[0116][0117][0118]
本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本发明内容、精神和范围内对本文 所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文 内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而 易见的,它们都被视为包括在本发明内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。