1.本发明涉及生物医药技术领域,尤其是一种基因编辑系统和方法,具体涉及一种用于外源基因定点插入的基因编辑系统和方法。
背景技术:
2.稳定表达细胞株又称稳转细胞株是指通过分子生物学方法,使特定基因质粒dna整合到细胞染色体上,使细胞长期稳定表达或干扰基因表达,它是基因功能研究、模型建立、作物育种、药物开发及基因治疗等最常用的手段之一。
3.传统稳转细胞株的构建方法主要可分为非病毒和病毒两大类,非病毒稳转细胞株的构建主要通过电转或脂质体等转染质粒或转座子到细胞中,利用抗生素(如嘌呤霉素)半数致死浓度对细胞不断加压,从而使包含特定基因的质粒整合到染色体dna,最终通过多轮细胞筛选从而获得稳转细胞株;病毒方法多为使用逆转录病毒和慢病毒等,按照一定的moi对目的细胞进行转染,转染48h或72h后对细胞进行筛选,获得稳定表达的细胞株。
4.但上述两种方法获得的稳定表达细胞株均发生了目的基因在染色体dna的随机整合,很容易引起其它基因功能的丧失,同时插入的拷贝数和位置不固定。因此,同一来源的细胞系中不同的细胞间表达差异比较明显,可能存在潜在的风险,干扰最终的实验结果,在免疫治疗中,这种随机整合也会带来潜在的致病、致癌风险。
技术实现要素:
5.针对上述目的,本发明的目的是提供一种用于外源基因定点插入的基因编辑系统和方法,从而使改造后的细胞遗传背景更为简单和统一,排除插入位点和拷贝数对其它基因功能的影响。
6.本发明一方面提供一种用于外源基因定点插入的基因编辑系统,包括cas9、sgrna和ssdna,所述cas9和sgrna构成rnp复合物,所述sgrna为外源基因的插入位点,所述ssdna为外源基因的插入模板。
7.在本发明的优选实施例中,所述插入位点包括pd1、tigit、aavs1、ctla-4、tim3和lag3。
8.在本发明的优选实施例中,当所述插入位点为aavs1时,所述sgrna的核苷酸序列如seq id no:1所示或其同源序列;所述sgrna的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上。
9.在本发明的优选实施例中,在本发明的优选实施例中,所述外源基因为aapc1,所述ssdna包括aapc1和设于其两侧的同源臂,所述aapc1的结构为ef1a-cd19-cd86-cd64-polya,所述两侧的同源臂分别为lha和rha,所述lha与ef1a相连,所述rha与polya相连。
10.在本发明的具体实施方式中,所述aapc1的核苷酸序列如seq id no:2所示或其同
源序列;所述aapc1的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上;和/或
11.所述lha的核苷酸序列如seq id no:3所示或其同源序列;所述lha的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上;和/或
12.所述rha的核苷酸序列如seq id no:4所示或其同源序列;所述rha的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上。
13.本发明一方面还提供一种用于外源基因定点插入的方法,包括以下步骤:
14.1)根据要插入的外源基因,选择插入位点,再根据所述插入位点设计、合成sgrna和ssdna;
15.2)然后将cas9和所述sgrna形成rnp复合物,然后再将所述rnp复合物与所述ssdna形成用于外源基因定点插入的基因编辑系统(rnp-ssdna复合物);
16.3)培养k562细胞,再将所述外源基因通过步骤2)中形成的用于外源基因定点插入的基因编辑系统插入所述k562细胞中,得到定点插入外源基因的k562细胞,即得。
17.在本发明的优选实施例中,在步骤1)中,所述插入位点包括pd1、tigit、aavs1、ctla-4、tim3和lag3。
18.在本发明的优选实施例中,当所述插入位点为aavs1时,所述sgrna的核苷酸序列如seq id no:1所示或其同源序列;所述sgrna的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上。
19.在本发明的优选实施例中,在步骤3)中,将所述外源基因通过电转或脂质体转染插入所述k562细胞中。
20.在本发明的优选实施例中,所述外源基因为aapc1,所述ssdna包括aapc1和设于其两侧的同源臂,所述aapc1的结构为ef1a-cd19-cd86-cd64-polya,所述两侧的同源臂分别为lha和rha,所述lha与ef1a相连,所述rha与polya相连。
21.在本发明的具体实施方式中,所述aapc1的核苷酸序列如seq id no:2所示或其同源序列;所述aapc1的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上;和/或
22.所述lha的核苷酸序列如seq id no:3所示或其同源序列;所述lha的同源序列与原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上;和/或
23.所述rha的核苷酸序列如seq id no:4所示或其同源序列;所述rha的同源序列与
原序列的同源性为95%或以上、96%或以上、97%或以上、98%或以上、99%或以上、99.1%或以上、99.2%或以上、99.3%或以上、99.4%或以上、99.5%或以上、99.6%或以上、99.7%或以上、99.8%或以上、或99.9%或以上。
24.本发明的有益效果是,主要利用基因编辑技术,通过ssdna作为外源基因的修复(插入)模板,在sgrna的配合下,成功在aavs1位点实现4.5kb(aapc1:ef1a-cd19-cd86-cd64-polya)的定点敲入;aavs1位点是位于人类基因ppp1r12c第一个内含子上的一段特定序列,在该位点中敲入外源基因片段具有稳定表达、不影响其他基因转录的优点;从而使改造后的细胞遗传背景更为简单和统一,排除插入位点和拷贝数对其它基因功能的影响。与传统稳转细胞株构建方法相比,本发明的方法构建的稳转细胞更加精确、安全,遗传背景更加清晰可靠,同时解决了大片段基因定点敲入细胞株构建难题。
附图说明
25.构成说明书的一部分的附图描述了本发明的实施例,并且连同描述一起用于解释本发明的原理。
26.参照附图,根据下面的详细描述,可以更加清楚地理解本发明,其中:
27.图1为本发明的实施例1中的目的基因组扩增的电泳结果图,其中1为marker,2为目的基因的扩增片段;
28.图2a为本发明的实施例1中的sgrna体外转录模板制备的电泳图,其中1为sgrna体外转录模板,2为marker;
29.图2b为本发明的实施例1中的sgrna效率验证的电泳图,其中,1为sgrna体外转录模板的扩增片段,2为sgrna体外转录模板的酶切片段,m为marker;
30.图3为本发明的实施例1中的外源基因的结构简图;
31.图4为本发明的实施例3中的aapc1定点敲入k562细胞电转效率检测结果;
32.图5为本发明的实施例4中的aapc1定点敲入的k562细胞的流式分选结果;
33.图6为本发明的实施例5中的aapc1定点敲入k562细胞前后aapc1中基因表达率检测结果;
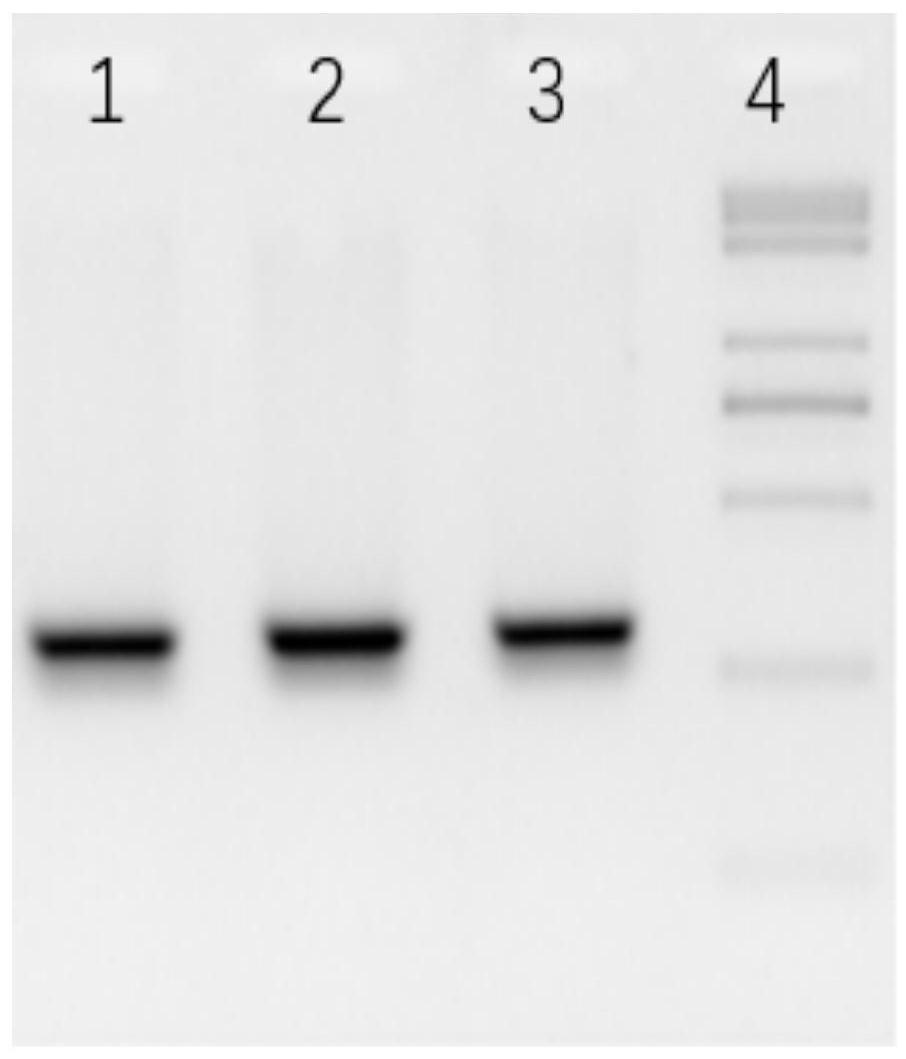
34.图7为本发明的实施例6中的aapc1定点敲入k562细胞亚克隆1-3(clone1-clone3)aapc1片段定点敲入基因组水平鉴定结果,其中,1-3为扩增的aavs1-p1片段,4-6为扩增的aavs1-p2片段,7为1kb的marker,8-10为扩增的aavs1-p3片段;
35.图8为本发明的实施例6中的k562细胞定点敲入aapc1基因组水平拷贝数鉴定结果,其中,1-3为亚克隆1-3(clone1-clone3),4为2kb的marker;
36.图9为本发明的实施例6中的aapc1基因定点敲入k562细胞的基因型结果,其中,k562-wt-seq为野生型k562基因组的序列,k562-ki-aapc1为aapc1基因定点敲入k562细胞(clone1-clone3)的基因型结果;
37.其中,k562-control为野生型k562,k562-rnp-ssdna为rnp-ssdna转染的k562细胞,k562-ki-aapc1为aapc1定点敲入的k562细胞。
具体实施方式
38.现在将参照附图来详细描述本发明的各种示例性实施例。
39.以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。
40.对于相关领域普通技术人员已知的技术、方法和设备可能不作详细讨论,但在适当情况下,所述技术、方法应当被视为说明书的一部分。
41.除非特别指明,本文中的“ssdna”即“single strand dna”,具体地,在本文中专指“外源基因的插入(修复)模板,包括外源基因和设于其两侧的同源臂”。
42.实施例1:外源基因和sgrna的设计、合成
43.1.sgrna的设计
44.利用crispr/cas9技术,针对aavs1区域,使用crisprgold网站(https://crisprgold.mdc-berlin.de/index.php)设计sgrna,选择的sgrna序列如seq id no:1(ggggccactagggacaggat)所示。
45.2.目的基因组扩增
46.取1
×
106细胞,使用天根细胞基因抽提试剂盒(dp304)抽提基因组dna,使用引物aavs1-f1(seq id no:5,gcctccccttcttgtaggcc)和aavs1-r1(seq id no:6,agccaaagttagaactcagg)进行基因组pcr扩增;再将扩增的片段使用凝胶试剂盒(axygen:ap-gx-250),切胶回收目的片段,结果如图1所示,并将回收的目的片段于-20℃保存,待用。
47.3.aavs1插入位点sgrna体外制备与编辑效率验证
48.sgrna体外转录:根据选择的sgrna序列,合成引物aavs1-sgrna-f(seq id no:7,taatacgactcactataggggggccactagggacaggattgttttagagctagaaatagcaagtt)aavs1-sgrna-r(seq id no:8,aaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctct),进行sgrna体外转录模板扩增,使用凝胶试剂盒(axygen:ap-gx-250),切胶回收目的片段,结果如图2a所示。
49.使用neb体外转录试剂盒(e2050s),按照说明书操作步骤进行体外转录合成sgrna,spcas9蛋白购自南京金斯瑞(z03393-100),以扩增的基因组为模板,37℃酶切30min,按照说明书进行体外编辑效率验证,结果如图2b所示,说明选择的sgrna体外具有较高的活性,可作为后续aapc1插入位点。
50.4.外源基因的设计、合成
51.选取sgrna所在位点作为aapc1的插入位点,插入位点两端分别选取约300-400bp作为同源臂(ha),左右两侧同源臂分别为lha(seq id no:3)和rha(seq id no:4),aapc1的结构为ef1a-cd19-cd86-cd64-polya-rhr(seq id no:2),外源基因(结构简图如图3所示)的全长序列委托苏州金唯智生物科技有公司限合成。
52.实施例2:外源基因定点插入k562细胞
53.1.k562细胞复苏培养
54.提前开启水浴锅,并设置温度为37℃,imdm-full培养基取出预热,从液氮罐中取出nk-92细胞,立即放入37℃水浴锅,取一支15ml离心管,加入8ml预热过的k562培养基;将细胞放在37℃水浴锅中不断轻轻晃动直至完全融化,用带酒精的无纺布擦拭冻存管外壁,将细胞悬液缓慢加至15ml离心管中(此过程控制在3min以内),混匀后1000rpm离心5min,弃上清;取1ml培养基重悬细胞沉淀,转移至含30ml imdm-full培养基的t75培养瓶中培养,按照一定密度进行细胞传代,保持合适的细胞生长密度。
55.2.k562细胞rnp-ssdna复合物电转
56.预先在t25培养瓶中加入10ml的imdm-full培养基,放置在培养箱中预热,从4℃冰箱中取出电转液至室温;再取适量培养的k562细胞至50ml离心管中,1500rpm离心5min,弃上清;然后加入20ml pbs重悬细胞沉淀,1500rpm离心5min,弃上清;再加入适量pbs重悬细胞沉淀,对细胞进行计数,按每管1
×
106细胞分装至1.5ml离心管中,2500rpm离心5min,弃尽上清。
57.电转前20min,用移液器分别吸取9μg spcas9和6μg实施例1制得的sgrna放于pcr管中,室温孵育10min后,加入3μg ssdna(由金唯智合成),混匀后室温放置5min,得到rnp-ssdna复合物(即用于外源基因定点插入的基因编辑系统)。
58.再取等量ab电转液重悬细胞沉淀后,加入孵育的rnp-ssdna复合物,使用celetrix(ctx-1500a-le)电转仪进行电转;电转结束后,得到外源基因定点插入的k562细胞,将所述k562细胞转移至预热的t25培养瓶中,放于37℃,5%co2培养箱中培养,得到aapc1定点敲入的k562细胞。
59.实施例3:aapc1定点敲入k562细胞电转效率检测
60.取实施例2中的部分转染24h后的aapc1定点敲入的k562细胞,转移至15ml离心管中,1500rpm离心5min,弃上清;加入1ml pbs清洗细胞沉淀后,转至1.5ml ep管,1500rpm离心5min,弃上清,重复操作一次后,使用流式(acea:novocyte3130),选取fitc通道进行电转效率检测,结果如图4所示,从图中可以看出rnp复合物电转k562细胞具有49.71%的转染效率。
61.实施例4:aapc1定点敲入k562细胞的分选
62.取实施例2中的转染5-7天后的aapc1定点敲入的k562细胞,转移至50ml离心管中,1500rpm离心5min,弃上清,得到细胞沉淀;然后加入20ml pbs清洗细胞沉淀,1500rpm离心5min,弃上清;再加入适量pbs重悬细胞沉淀,加入适量anti-cd19抗体(biolegend:货号392503)避光染色20min;染色结束后,加入20ml pbs清洗细胞,1500rpm离心5min,弃上清;再加入20ml pbs清洗细胞沉淀,1500rpm离心5min,弃上清;然后加入适量pbs 0.2%fbs重悬细胞沉淀,细胞悬液用40μm滤网过滤后用于第一轮流式分选,分选后的细胞转移至t25培养瓶中培养,待细胞长起来后,进行流式第二轮流式分选,步骤同上,电转前后流式分选结果如图5所示,从图5可以看出aapc1定点敲入k562细胞的敲入效率约为0.99%,经两轮细胞分选后,获得纯度99%以上aapc1定点敲入的k562细胞。
63.实施例5:单克隆细胞筛选
64.选取实施例4中第二轮分选的细胞进行亚克隆96孔板种板,待细胞长起来后,选择3株亚克隆细胞(clone1-clone3)用于后续基因过表达及基因组水平敲入鉴定。
65.取上述亚克隆细胞和未转染的k562细胞(对照)进行cd19,cd64,cd86细胞阳性表达率检测,使用抗体为anti-cd19(biolegend,货号:392503),anti-cd64(biolegend,货号:305005),anti-cd86(biolegend,货号:374203),细胞处理方式参照相应说明书进行,检测结果如图6所示,表明定点敲入aapc1的k562细胞cd19,cd64,cd86均具有很高表达率,成功获得稳定表达aapc1的k562细胞。
66.实施例6:基因组水平敲入鉴定
67.从clone1-clone3选取1
×
106数量的细胞,按照细胞基因组抽提试剂盒说明书抽
提基因组,设计三组嵌套引物别为aavs1-f2(seq id no:9,cctgtgccatctctcgtttct),aavs1-r2(seq id no:10,gcctgggaatccacatgagg);aavs1-f3(seq id no:11,aggaacctctagtggtgaaggt),aavs1-r3(seq id no:12,acgagaagaaggtcatcagca);aavs1-f4(seq id no:13,acacccgtctggtttcacg),aavs1-r4(seq id no:14,caagaggagaagcagtttgga),进行pcr扩增,对定点敲入的片段及位点进行检测,其对应的克隆片段分别如图3所示,第一段(aavs1-p1)位于aavs1-f2到aavs1-r2之间,第二段(aavs1-p2)位于aavs1-f3到aavs1-r3之间,第三段(aavs1-p3)位于aavs1-f4到aavs1-r4之间;pcr扩增结果如图7所示,从图中可以看出亚克隆clone1-clone3成功扩增出aapc1片段。对上述步骤中的pcr产物按照凝胶回收试剂盒操作步骤回收目的片段后进行sanger测序分析,结果表明4.5kb片段成功敲入到aavs1位点。
68.分别使用aavs1-f5(seq id no:15,ggtccgagagctcagctagt),aavs1-r5(seq id no:16,acaggaggtgggggttaga)引物对clone1-clone3细胞株进行基因组水平拷贝数鉴定,结果如图8所示,从图中可以看出,其扩增条带大小为275bp,表明lhr-ef1a-cd19-cd64-cd86-polya-rha为单拷贝敲入;此外,对clone1-clone3进行测序,其基因型如图9所示,说明clone1-clone3细胞株的基因型为单拷贝定点敲入。
69.综上,本发明主要利用基因编辑技术,通过ssdna作为外源基因的修复(插入)模板,在sgrna的配合下,成功在aavs1位点实现4.5kb(aapc1:ef1a-cd19-cd86-cd64-polya)的定点敲入,并且通过实验证明,该外源基因敲入为单拷贝敲入;从而使改造后的细胞遗传背景更为简单和统一,排除插入位点和拷贝数对其它基因功能的影响。与传统稳转细胞株构建方法相比,该方法构建的稳转细胞更加精确、安全,遗传背景更加清晰可靠,同时解决了大片段基因定点敲入细胞株构建难题。
70.本发明的描述是为了示例和描述起见而给出的,而并不是无遗漏的或者将本发明限于所公开的形式。很多修改和变化对于本领域的普通技术人员而言是显然的。选择和描述实施例是为了更好说明本发明的原理和实际应用,并且使本领域的普通技术人员能够理解本发明从而设计适于特定用途的带有各种修改的各种实施例。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。