1.本发明属于电池材料技术领域,具体涉及一种双草酸磷酸盐的制备方法,一种双草酸磷酸盐衍生物及其制备方法,一种电解质盐,一种电解液,以及一种二次电池。
背景技术:
2.随着社会需求的不断发展,离子电池的使用寿命、高低温性能、安全性能、倍率性能等已不能满足动力电池发展的要求。提升动力电池性能有多种途径,其中离子电池电解质盐的结构、性质对于离子电池的电化学性能起着至关重要的作用。到目前为止,人们已开发出数目众多的新型离子电池的电解质盐,这些盐虽然具有更好的热稳定性和高低温性能,但也有一些明显的不足,如溶解度低、合成难度大、价格昂贵、腐蚀集流体等。
3.其中,二氟双草酸磷酸锂(lidodfp)是近年来开发的一种新型电解质锂盐,与六氟磷酸锂相比,lidodfp具有更好的热稳定性和对水的耐受性,同时还可以在正极材料表面形成更加稳定的固体电解质界面膜(sei膜),有效提高电池的高温循环和高温存储性能,因此在高镍、高电压领域有着广泛的应用。然而,现有的lidodfp产品多为二氟双草酸磷酸锂溶液,存在氯离子浓度和游离酸含量偏高的问题,限制了其在锂离子二次电池电解液中的应用。因此,寻找一种无水双草酸磷酸盐的制备方法是当前新型电解质盐的研究重点之一。
技术实现要素:
4.本发明的目的是提供一种双草酸磷酸盐的制备方法,一种双草酸磷酸盐衍生物及其制备方法,一种电解质盐,一种电解液,以及一种二次电池,旨在解决现有双草酸磷酸盐的制备方法中存在的产品纯度较低的技术问题。
5.为了实现上述发明目的,本发明采用的技术方案如下:
6.本发明一方面提供了一种双草酸磷酸盐的制备方法,其包括如下步骤:
7.提供草酸、卤代硅烷化合物、有机碱、六氟磷酸盐和第一非水溶剂,所述六氟磷酸盐的化学通式为mpf6,且m为li、na或k;
8.在所述第一非水溶剂中,将所述草酸、所述卤代硅烷化合物和所述有机碱进行混合反应,得到草酸硅基酯溶液;
9.所述草酸硅基酯溶液与所述六氟磷酸盐按照(2-2.2:1)的摩尔比进行混合反应后,得到所述双草酸磷酸盐;所述双草酸磷酸盐为二氟双草酸磷酸盐,其结构式如式(i)所示,且m为li、na或k:
10.11.作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,所述卤代硅烷化合物的结构式如式(ii)所示:
[0012][0013]
其中,r1、r2、r3分别独立地选自氢原子、碳原子数为1-10的烷基、碳原子数为2-10的烯基、碳原子数为2-10的炔基、碳原子数为1-10的烷氧基、碳原子数为6-20的芳香基团中的一种,且x为cl、br、i或f。
[0014]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,将所述草酸、所述卤代硅烷化合物和所述有机碱进行混合反应的步骤中,所述草酸、所述卤代硅烷化合物与所述有机碱的摩尔比为1:(2-3):(2-4)。
[0015]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,将所述草酸、所述卤代硅烷化合物和所述有机碱进行混合反应的步骤中,所述混合反应的反应温度为-20℃~20℃,反应时间为1h-6h。
[0016]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,所述草酸硅基酯溶液与所述六氟磷酸盐进行混合反应的步骤中,所述混合反应的反应温度为40℃~80℃,反应时间为1h-3h。
[0017]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,所述草酸的水分含量小于等于100ppm。
[0018]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,所述有机碱选自三乙胺、二异丙基乙胺、三异丙基胺、三丙胺、吡啶、2-甲基吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶、吗啉、n-甲基吗啉、n-乙基吗啉、哌啶、n-甲基哌啶、n-乙基哌啶、吡嗪、n-甲基吡嗪、n,n'-二甲基吡嗪、n-乙基吡嗪、n,n'-二乙基吡嗪、哌嗪、n-甲基哌嗪、n,n'-二甲基哌嗪、n-乙基哌嗪、n,n'-二乙基哌嗪、咪唑、n-甲基咪唑、n-乙基咪唑、1,8-二氮杂二环十一碳-7-烯、1,5-二氮杂双环[4.3.0]壬-5-烯、三氮脒、胍、四甲基胍中的至少一种。
[0019]
作为本发明双草酸磷酸盐的制备方法的一种优选技术方案,所述第一非水溶剂选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0020]
本发明另一方面提供一种双草酸磷酸盐衍生物的制备方法,其包括如下步骤:
[0021]
提供二氟双草酸磷酸盐、硅基化合物和第二非水溶剂,所述硅基化合物的结构中含氰基或异氰酸酯基,所述二氟双草酸磷酸盐的结构式如式(i)所示,且m为li、na或k:
[0022][0023]
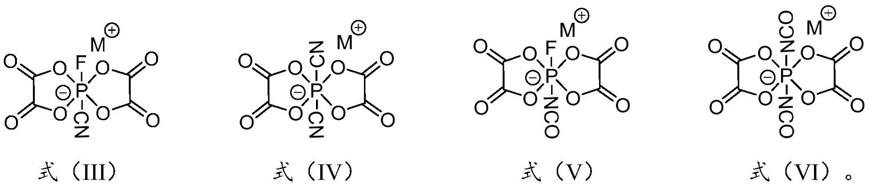
在所述第二非水溶剂中,将所述二氟双草酸磷酸盐与所述硅基化合物进行混合反应,得到所述双草酸磷酸盐衍生物;所述双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐、双草酸二异氰酸酯磷酸盐中的至少一种,所述双草酸氰基氟磷酸盐、所述双草酸二氰基磷酸盐、所述双草酸异氰酸酯氟磷酸盐、所述双草酸二异氰酸酯磷酸盐的结构式依次如式(iii)-(vi)所示,且m为li、na或k:
[0024][0025]
作为本发明双草酸磷酸盐衍生物的制备方法的一种优选技术方案,所述二氟双草酸磷酸盐通过本发明所述双草酸磷酸盐的制备方法制备得到。
[0026]
作为本发明双草酸磷酸盐衍生物的制备方法的一种优选技术方案,所述硅基化合物为三甲基硅氰化物或三甲基硅异氰酸酯化物。
[0027]
作为本发明双草酸磷酸盐衍生物的制备方法的一种优选技术方案,将所述二氟双草酸磷酸盐与所述硅基化合物进行混合反应的步骤中,所述二氟双草酸磷酸盐与所述硅基化合物的摩尔比为1:(1-3)。
[0028]
作为本发明双草酸磷酸盐衍生物的制备方法的一种优选技术方案,将所述二氟双草酸磷酸盐与所述硅基化合物进行混合反应的步骤中,所述混合反应的反应温度为0℃~60℃,反应时间为1h-6h。
[0029]
作为本发明双草酸磷酸盐衍生物的制备方法的一种优选技术方案,所述第二非水溶剂选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0030]
本发明再一方面提供了一种双草酸磷酸盐衍生物,所述双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐或双草酸二异氰酸酯磷酸盐,所述双草酸氰基氟磷酸盐、所述双草酸二氰基磷酸盐、所述双草酸异氰酸酯氟磷酸盐、所述双草酸二异氰酸酯磷酸盐的结构式依次如式(iii)-(vi)所示,且m为li、na或k:
[0031][0032]
本发明又一方面提供了一种电解质盐,其包括上述双草酸磷酸盐的制备方法制备得到的双草酸磷酸盐,或上述双草酸磷酸盐衍生物的制备方法制备得到的双草酸磷酸盐衍生物,或上述的双草酸磷酸盐衍生物。
[0033]
本发明还一方面提供了一种电解液,其包括上述的电解质盐。
[0034]
本发明最后一方面提供一种二次电池,其包括上述的电解液。
[0035]
本发明提供的双草酸磷酸盐的制备方法具有以下优点:
[0036]
首先,通过将草酸、卤代硅烷化合物和有机碱进行混合反应,卤代硅烷化合物可以使原本在非水溶剂中的溶解度较差的草酸逐渐溶解,为两者的反应提供良好的反应环境;有机碱不仅可促进卤代硅烷化合物与草酸之间的反应进程,提升反应速率,还可去除反应产生的酸性气体及多余草酸,以提高双草酸磷酸盐产品的纯度。
[0037]
其次,该方法为两步反应法,先利用草酸、卤代硅烷化合物和有机碱反应生成中间产物草酸硅基酯,草酸硅基酯再与六氟磷酸盐反应得到双草酸磷酸盐。该方法可以促进草酸与卤代硅烷化合物完全反应,使双草酸磷酸盐产品更加纯净、得率更高。更重要的是,本发明提供的制备方法中,所有的反应物均为有机物,溶剂为非水溶剂,因此可通过浓缩干燥得到纯度高的双草酸磷酸盐,避免了氯离子浓度和游离酸偏高的问题,且反应过程中的原子经济性高、杂质少,无需事先合成反应原料,在简化反应过程、节约生产成本的同时也提升了反应过程的安全性,对环境更加友好。
[0038]
此外,本发明提供的双草酸磷酸盐的制备方法还可作为制备一系列双草酸磷酸盐衍生物的前步骤,实现了类似方法制备多种产品、多种产品可以使用同一套仪器设备生产的效果,减少了设备投入、劳动力成本和能耗,具有良好的工业化应用前景。
[0039]
本发明提供的双草酸磷酸盐衍生物的制备方法中,以二氟双草酸磷酸盐为原料,通过在非水溶剂中与含氰基或异氰酸酯基的硅基化合物进行反应得到双草酸磷酸盐衍生物。该制备方法中的反应物均为有机物,溶剂为非水溶剂,因此可通过浓缩干燥得到纯度高的双草酸磷酸盐衍生物,避免了氯离子浓度和游离酸偏高的问题,且原料成本低,反应过程中的原子经济性高、杂质少,简化了反应过程,节约了生产成本,还提升了反应过程的安全性,对环境更加友好。
[0040]
本发明提供的双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐或双草酸二异氰酸酯磷酸盐,其是四种新型的双草酸磷酸盐,水分低于20ppm,酸度低于50ppm,氯离子浓度低于5ppm,具有较好的热稳定性和较高的离子电导率,应用前景良好。
[0041]
本发明提供的电解质盐,包括上述的双草酸磷酸盐及其衍生物的制备方法制备得到的双草酸磷酸盐及其衍生物,或包括上述的双草酸磷酸盐衍生物。由于上述制备方法中的反应物均为有机物,溶剂为非水溶剂,且所得双草酸磷酸盐及其衍生物纯度高,水分低于20ppm,酸度低于50ppm,氯离子浓度低于5ppm,具有较好的热稳定性和较高的离子电导率,
因此将其作为电解质盐时,可以有效抑制电解液在存储过程中的水分和酸度的上升,提升电解液的稳定性和安全性。
[0042]
本发明提供的电解液,包括上述的电解质盐。由于该电解质盐可以有效抑制电解液在存储过程中的水分和酸度的上升,所以本发明提供的电解液具有良好的稳定性和安全性。
[0043]
本发明提供的二次电池,包括上述的电解液。该电解液可以有效改善所得二次电池的安全性和稳定性,经实验证明,本发明提供的二次电池在常温、高温下均具有良好的循环性能和存储性能,使用寿命更长。
具体实施方式
[0044]
为使本发明实施例的目的、技术方案和技术效果更加清楚,对本发明实施例中的技术方案进行清楚、完整地描述,以下所描述的实施例是本发明一部分实施例,而不是全部的实施例。结合本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行;所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0045]
在本发明的描述中,术语“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b的情况。其中a,b可以是单数或者复数。字符“/”一般表示前后关联对象是一种“或”的关系。
[0046]
在本发明的描述中,“至少一个”是指一个或者多个,“多个”是指两个或两个以上。“以下至少一项(个)”或其类似表达,是指的这些项中的任意组合,包括单项(个)或复数项(个)的任意组合。例如,“a,b,或c中的至少一项(个)”,或,“a,b,和c中的至少一项(个)”,均可以表示:a、b、c、a-b(即a和b)、a-c、b-c、或a-b-c,其中a、b、c分别可以是单个,也可以是多个。
[0047]
需要理解的是,本发明实施例中所提到的相关成分的重量不仅仅可以指代各组分的具体含量,也可以表示各组分间重量的比例关系,因此,只要是按照本发明实施例相关组分的含量按比例放大或缩小均在本发明公开的范围之内。具体地,本发明实施例中所述的重量可以是μg、mg、g、kg等化工领域公知的质量单位。
[0048]
另外,除非上下文另外明确地使用,否则词的单数形式的表达应被理解为包含该词的复数形式。术语“包括”或“具有”旨在指定特征、数量、步骤、操作、元件、部分或者其组合的存在,但不用于排除存在或可能添加一个或多个其它特征、数量、步骤、操作、元件、部分或者其组合。
[0049]
本发明实施例提供了一种双草酸磷酸盐的制备方法,其包括如下步骤:
[0050]
s1、提供草酸、卤代硅烷化合物、有机碱、六氟磷酸盐和第一非水溶剂,六氟磷酸盐的化学通式为mpf6,且m为li、na或k;
[0051]
s2、在第一非水溶剂中,将草酸、卤代硅烷化合物和有机碱进行混合反应,得到草酸硅基酯溶液;
[0052]
s3、草酸硅基酯溶液与六氟磷酸盐按照(2-2.2):1的摩尔比进行混合反应后,得到双草酸磷酸盐;
[0053]
其中,所得的双草酸磷酸盐为二氟双草酸磷酸盐(mdodfp),其结构式如式(i)所示,且m为li、na或k:
[0054][0055]
本发明实施例提供的双草酸磷酸盐的制备方法具有以下优点:
[0056]
首先,通过将草酸、卤代硅烷化合物和有机碱进行混合反应,卤代硅烷化合物可以使原本在非水溶剂中的溶解度较差的草酸逐渐溶解,为两者的反应提供良好的反应环境;有机碱不仅可促进卤代硅烷化合物与草酸之间的反应进程,提升反应速率,还可去除反应产生的酸性气体及多余草酸,以提高双草酸磷酸盐产品的纯度。
[0057]
其次,该方法为两步反应法,先利用草酸、卤代硅烷化合物和有机碱反应生成中间产物草酸硅基酯,草酸硅基酯再与六氟磷酸盐反应得到双草酸磷酸盐。该方法可以促进草酸与卤代硅烷化合物完全反应,使双草酸磷酸盐产品更加纯净、得率更高。更重要的是,本发明提供的制备方法中,所有的反应物均为有机物,溶剂为非水溶剂,因此可通过浓缩干燥得到纯度高的双草酸磷酸盐,避免了氯离子浓度和游离酸偏高的问题,且反应过程中的原子经济性高、杂质少,无需事先合成反应原料,在简化反应过程、节约生产成本的同时也提升了反应过程的安全性,对环境更加友好。
[0058]
此外,本发明实施例提供的双草酸磷酸盐的制备方法还可作为制备一系列双草酸磷酸盐衍生物的前步骤,实现了类似方法制备多种产品、多种产品可以使用同一套仪器设备生产的效果,减少了设备投入、劳动力成本和能耗,具有良好的工业化应用前景。
[0059]
具体地,s1中,草酸在本发明实施例中是中间产物草酸硅基酯溶液的反应原料之一。在一些实施例中,为了进一步减少反应体系的水分,减少游离酸,选择水分含量小于等于100ppm的草酸。在一些实施例中,对草酸原料进行前处理,得到水分含量小于等于100ppm的草酸。在一些具体实施例中,可以采用如下方法进行前处理:在温度为40℃-100℃的真空条件下,将草酸原料进行干燥,直至草酸的水分含量小于等于100ppm。
[0060]
卤代硅烷化合物是中间产物草酸硅基酯溶液的又一反应原料,卤代硅烷化合物与草酸在含有机碱的环境中反应,得到中间产物草酸硅基酯。本技术实施例采用卤代硅烷化合物与草酸反应制备中间产物草酸硅基酯,卤代硅烷化合物可以促进草酸完全溶解,进而与草酸和有机碱发生反应。在一些实施例中,卤代硅烷化合物的结构式如式(ii)所示:
[0061][0062]
其中,r1、r2、r3分别独立地选自氢原子、碳原子数为1-10的烷基、碳原子数为2-10的烯基、碳原子数为2-10的炔基、碳原子数为1-10的烷氧基、碳原子数为6-20的芳香基团中的一种,且x为cl、br、i或f。在一些具体实施例中,x为cl、br或i,以避免生成hf有害气体。
[0063]
在一些实施例中,卤代硅烷化合物的结构式中x为cl,r1、r2、r3三个取代基团分别独立地选自烷基、烯基、炔基、烷氧基、氨基、烷基氨基或芳香基团。在一些具体实施例中,r1、r2、r3均为甲基,或r1为叔丁基、r2、r3均为甲基,或r1为二甲基氨基、r2、r3均为甲基。其中,当r1、r2、r3均为甲基时,生产成本最低;当r1为叔丁基、r2、r3均为甲基时,中间产物草酸硅基酯的稳定性更好,对提高产率有帮助;当r1为二甲基氨基、r2、r3均为甲基时,对降低产物的水分有帮助。
[0064]
有机碱,用于促进卤代硅烷化合物与草酸发生反应,也用于去除草酸和卤代硅烷化合物反应生成的酸性气体和反应体系中多余的草酸等杂质,使草酸等杂质以沉淀的方式去除,提升双草酸磷酸盐产品的纯度。在一些实施例中,有机碱选自三乙胺、二异丙基乙胺、三异丙基胺、三丙胺、吡啶、2-甲基吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶、吗啉、n-甲基吗啉、n-乙基吗啉、哌啶、n-甲基哌啶、n-乙基哌啶、吡嗪、n-甲基吡嗪、n,n'-二甲基吡嗪、n-乙基吡嗪、n,n'-二乙基吡嗪、哌嗪、n-甲基哌嗪、n,n'-二甲基哌嗪、n-乙基哌嗪、n,n'-二乙基哌嗪、咪唑、n-甲基咪唑、n-乙基咪唑、1,8-二氮杂二环十一碳-7-烯、1,5-二氮杂双环[4.3.0]壬-5-烯、三氮脒、胍、四甲基胍中的至少一种。三乙胺和吡啶的价格低廉,是最常用的有机碱,然而吡啶的毒性较大,因此从环保和降低生产成本的角度优选三乙胺。
[0065]
第一非水溶剂是不含水的溶剂。由于使用含水的溶剂制备得到的是双草酸磷酸盐溶液,难以通过结晶析出纯度高的双草酸磷酸盐,且存在氯离子浓度和游离酸偏高的问题,因此本发明实施例采用非水溶剂来克服上述问题。在一些实施例中,第一非水溶剂选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0066]
s2中,草酸、卤代硅烷化合物和有机碱(c)进行混合反应是在第一非水溶剂体系中进行,反应式如下:
[0067][0068]
其中,为了便于控制反应物的添加量和反应的发生,在一些实施例中,可将草酸先与第一非水溶剂混合,由于草酸的溶解性较差,此时仅发生了部分溶解,体系为悬浊液;然后在搅拌条件下向悬浊液中加入卤代硅烷化合物的非水溶液,使体系中的固体完全溶解形成均一的溶液体系,同时有酸性气体放出(当卤代硅烷化合物中的x为cl时,生成的酸性气体为hcl),反应释放的酸性气体可通过无机碱水溶液吸收。然后在均一的溶液体系中加入有机碱的非水溶液,此时会生成大量有机盐沉淀,过滤去除沉淀后,所得溶液即为草酸硅基酯溶液。
[0069]
进一步地,用于吸收酸性气体的无机碱水溶液中的无机碱选自氢氧化钠、氢氧化钾、碳酸钠、碳酸氢钠、碳酸钾中的至少一种,优选氢氧化钠的饱和水溶液,具有成本低、原
料易得、吸收完全的优点。
[0070]
进一步地,为了促进反应物的溶解和反应的发生,草酸与第一非水溶剂混合是在室温下进行;加入卤代硅烷化合物的非水溶液是在0℃-20℃下进行。
[0071]
在一些实施例中,草酸、卤代硅烷化合物和有机碱进行混合反应的步骤中,将三者的摩尔比控制在1:(2-3):(2-4),使反应物完全反应、减少多余杂质的产生。具体地,三者典型而非限制性的摩尔比为1:2:2、1:2:3、1:2:4、1:3:2、1:3:3、1:3:4。
[0072]
在一些实施例中,草酸、卤代硅烷化合物和有机碱进行混合反应的步骤中,将混合反应的温度控制在-20℃~20℃,反应时间控制在1h-6h,有利于避免反应过于剧烈,同时吸收反应释放的热量。具体地,典型而非限制性的混合反应温度为-20℃、-15℃、-10℃、-5℃、0℃、5℃、10℃、15℃、20℃;典型而非限制性的混合反应时间为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h。
[0073]
s3中,将中间产物草酸硅基酯溶液与六氟磷酸盐按照(2-2.2):1的摩尔比进行混合反应,得到双草酸磷酸盐,其反应式如下:
[0074][0075]
在一些实施例中,将草酸硅基酯溶液与六氟磷酸盐进行混合反应的温度控制在40℃~80℃,反应时间控制在1h-3h,以提升反应速率,促进反应完全。具体地,典型而非限制性的混合反应温度为40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃;典型而非限制性的混合反应时间为1h、1.5h、2h、2.5h、3h。
[0076]
草酸硅基酯溶液与六氟磷酸盐进行混合反应后,所得二氟双草酸磷酸盐为溶液态,在一些实施例中,还包括对该溶液进行浓缩干燥的步骤,以提升二氟双草酸磷酸盐的纯度。在一些实施例中,对该溶液进行浓缩干燥的步骤包括:先将二氟双草酸磷酸盐溶液室温减压蒸馏浓缩得到白色固体,然后将白色固体使用非水溶剂进行重结晶,得到白色晶体,再将白色晶体真空干燥,得到二氟双草酸磷酸盐。
[0077]
进一步地,将真空干燥的温度控制在30℃-100℃,优选40℃-80℃,真空干燥时间为1h-8h,优选3h-5h,以提高真空干燥的效率,使晶体充分干燥。具体地,典型而非限制性的真空干燥温度为30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、100℃;典型而非限制性的真空干燥时间为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、6.5h、7h、7.5h、8h。
[0078]
在一些实施例中,为了提升反应体系中各反应原料的溶解度,草酸、卤代硅烷化合物和有机碱均事先用第一非水溶剂溶解,此时所需第一非水溶剂的总质量为草酸的质量的1-10倍。
[0079]
相应地,本发明实施例还提供了一种双草酸磷酸盐衍生物的制备方法,其包括如下步骤:
[0080]
s4、提供二氟双草酸磷酸盐、硅基化合物和第二非水溶剂,硅基化合物的结构中含氰基或异氰酸酯基,二氟双草酸磷酸盐的结构式如式(i)所示,且m为li、na或k:
[0081][0082]
s5、在第二非水溶剂中,将二氟双草酸磷酸盐与硅基化合物进行混合反应,得到双草酸磷酸盐衍生物;所得双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐、双草酸二异氰酸酯磷酸盐中的至少一种,双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐、双草酸二异氰酸酯磷酸盐的结构式依次如式(iii)-(vi)所示,且m为li、na或k:
[0083][0084]
本发明实施例提供的双草酸磷酸盐衍生物的制备方法中,以二氟双草酸磷酸盐为原料,通过在非水溶剂中与含氰基或异氰酸酯基的硅基化合物进行反应得到双草酸磷酸盐衍生物。该制备方法中的反应物均为有机物,溶剂为非水溶剂,因此可通过浓缩干燥得到纯度高的双草酸磷酸盐衍生物,避免了氯离子浓度和游离酸偏高的问题,且原料成本低,反应过程中的原子经济性高、杂质少,简化了反应过程,节约了生产成本,还提升了反应过程的安全性,对环境更加友好。
[0085]
具体地,s4中,二氟双草酸磷酸盐可以采用常规方法制备得到,优选使用本发明实施例提供的上述双草酸磷酸盐的制备方法(步骤s1至步骤s3)制备得到的二氟双草酸磷酸盐,这是因为本发明实施例提供的上述双草酸磷酸盐的制备方法中所有的反应物均为有机物,溶剂为非水溶剂,所得二氟双草酸磷酸盐的纯度高,杂质少,且避免了氯离子浓度和游离酸偏高的问题。
[0086]
第二非水溶剂,在本发明实施例中作为二氟双草酸磷酸盐和硅基化合物的溶剂,由于其中不含有水,更有利于通过结晶析出纯度高的双草酸磷酸盐衍生物,且避免了游离酸偏高的问题。在一些实施例中,第二非水溶剂选自选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0087]
硅基化合物含有氰基或异氰酸酯基,用于与二氟双草酸磷酸盐发生反应,生成双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐、双草酸二异氰酸酯磷酸盐。在一些实施例中,硅基化合物为三甲基硅氰化物(me3sicn)或三甲基硅异氰酸酯化物(me3sinco)。其中,硅基化合物为me3sicn时,所得双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐和/或双草酸二氰基磷酸盐;硅基化合物为me3sinco时,所得双
草酸磷酸盐衍生物为双草酸异氰酸酯氟磷酸盐和/或双草酸二异氰酸酯磷酸盐。
[0088]
s5中,在第二非水溶剂中,二氟双草酸磷酸盐与硅基化合物进行混合反应,得到双草酸磷酸盐衍生物。在一些实施例中,二氟双草酸磷酸盐与硅基化合物的摩尔比为1:(1-3)时,使反应物完全反应、减少多余杂质的产生。具体地,典型而非限制性的二氟双草酸磷酸盐与硅基化合物的摩尔比为1:1、1:1.1、1:1.2、1:1.3、1:1.4、1:1.5、1:1.6、1:1.7、1:1.8、1:1.9、1:2、1:2.1、1:2.2、1:2.3、1:2.4、1:2.5、1:2.6、1:2.7、1:2.8、1:2.9、1:3。
[0089]
二氟双草酸磷酸盐与硅基化合物进行混合反应过程中,硅基化合物的含量不同,得到的产物也会存在差异。因此,本技术实施例可以通过调整二氟双草酸磷酸盐与硅基化合物的摩尔比,控制所得产物的种类。
[0090]
在一些实施例中,当硅基化合物为me3sicn,且二氟双草酸磷酸盐与me3sicn的摩尔比为1:1时,所得反应产物为双草酸氰基氟磷酸盐,其反应式如下所示:
[0091][0092]
在一些实施例中,当硅基化合物为me3sicn,且二氟双草酸磷酸盐与me3sicn的摩尔比为1:2时,所得反应产物为双草酸二氰基磷酸盐,反应式如下所示:
[0093][0094]
在一些实施例中,当硅基化合物为me3sicn,且二氟双草酸磷酸盐与me3sicn的摩尔比为1:(1-3)时,所得反应产物可能是双草酸氰基氟磷酸盐和双草酸二氰基磷酸盐的混合物。
[0095]
在一些实施例中,当硅基化合物为me3sinco,且二氟双草酸磷酸盐与me3sinco的摩尔比为1:1时,所得反应产物为双草酸异氰酸酯氟磷酸盐,反应式如下所示:
[0096][0097]
在一些实施例中,当硅基化合物为me3sinco,且二氟双草酸磷酸盐与me3sinco的摩尔比为1:2时,所得反应产物为双草酸二异氰酸酯磷酸盐,反应式如下所示:
[0098][0099]
为了便于控制反应物的添加量和反应的发生,在一些实施例中,可将二氟双草酸磷酸盐和硅基化合物分别先与第二非水溶剂混合溶解得到各自的非水溶液,然后再将两者
混合进行反应。该反应过程会有me3sif气体放出,可使用无机碱水溶液吸收;滴加完成后,静置过滤除去悬浮固体物质,得到呈无色透明的溶液,将该溶液进行浓缩干燥,得到双草酸磷酸盐衍生物。
[0100]
进一步地,用于吸收反应过程中放出的me3sif气体的无机碱水溶液中,无机碱选自氢氧化钠、氢氧化钾、碳酸钠、碳酸氢钠、碳酸钾中的至少一种,优选氢氧化钠的饱和水溶液,具有成本低、原料易得、吸收完全的优点。
[0101]
在一些实施例中,二氟双草酸磷酸盐与硅基化合物进行混合反应的步骤中,将混合反应的温度控制在0℃-60℃,反应时间控制在1h-6h,有利于反应的发生以及反应物完全反应。具体地,典型而非限制性的混合反应温度为0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃;典型而非限制性的混合反应时间为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h。
[0102]
由于二氟双草酸磷酸盐与硅基化合物进行混合反应后,所得双草酸磷酸盐衍生物为溶液态,在一些实施例中,为了提高双草酸磷酸盐衍生物的纯度,还包括对双草酸磷酸盐衍生物溶液进行浓缩干燥的步骤。在一些实施例中,对该溶液进行浓缩干燥的步骤包括:先将双草酸磷酸盐衍生物溶液室温减压浓缩得到淡黄色或白色固体,然后将该淡黄色或白色固体使用非水溶剂进行重结晶,得到白色晶体,再将白色晶体真空干燥,得到双草酸磷酸盐衍生物。
[0103]
进一步地,将真空干燥的温度控制在30℃-100℃,优选40℃-80℃,真空干燥时间为1h-8h,优选3h-5h,以提高真空干燥的效率,使晶体充分干燥。具体地,典型而非限制性的真空干燥温度为30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、100℃;典型而非限制性的真空干燥时间为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、6.5h、7h、7.5h、8h。
[0104]
相应地,本发明实施例提供了一种双草酸磷酸盐衍生物,其是双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐或双草酸二异氰酸酯磷酸盐,双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐、双草酸二异氰酸酯磷酸盐的结构式依次如式(iii)-(vi)所示,且m为li、na或k;
[0105][0106]
本发明实施例提供的双草酸磷酸盐衍生物为双草酸氰基氟磷酸盐、双草酸二氰基磷酸盐、双草酸异氰酸酯氟磷酸盐或双草酸二异氰酸酯磷酸盐,其是四种新型的双草酸磷酸盐,水分低于20ppm,酸度低于50ppm,氯离子浓度低于5ppm,具有较好的热稳定性和较高的离子电导率,应用前景良好。
[0107]
相应地,本发明实施例还提供了一种电解质盐,其包括上述双草酸磷酸盐的制备方法制备得到的双草酸磷酸盐,或上述双草酸磷酸盐衍生物的制备方法制备得到的双草酸磷酸盐衍生物,或上述的双草酸磷酸盐衍生物。
[0108]
本发明实施例提供的电解质盐,包括上述的双草酸磷酸盐及其衍生物的制备方法
制备得到的双草酸磷酸盐及其衍生物,或包括上述的双草酸磷酸盐衍生物。由于上述制备方法中的反应物均为有机物,溶剂为非水溶剂,且所得双草酸磷酸盐及其衍生物纯度高,水分低于20ppm,酸度低于50ppm,氯离子浓度低于5ppm,具有较好的热稳定性和较高的离子电导率,因此将其作为电解质盐时,可以有效抑制电解液在存储过程中的水分和酸度的上升,提升电解液的稳定性和安全性。
[0109]
相应地,本发明实施例还提供了一种电解液,其包括上述的电解质盐。
[0110]
本发明实施例提供的电解液,包括上述的电解质盐。由于该电解质盐可以有效抑制电解液在存储过程中的水分和酸度的上升,所以本发明实施例提供的电解液具有良好的稳定性和安全性。
[0111]
在一些实施例中,以电解液的总质量为100%计,本发明实施例提供的该电解质盐的含量为1%-15%。通过添加该含量的电解质盐,可以与电解液中的其它成分起到功能互补的作用,既可提高电解液的稳定性和安全性,又可确保电解液的电化学性能不会受较大影响。
[0112]
在一些实施例中,电解液包括六氟磷酸锂、非水溶剂和上述电解质盐,其中六氟磷酸锂与上述电解质盐配合使用。该电解液中的非水溶剂为碳酸酯类溶剂,其中碳酸酯为链状或环状的碳酸酯。在一些具体实施例中,环状酯选自碳酸乙烯酯(ec)、碳酸丙烯酯(vc)、γ-丁内酯、1,3-丙磺酸内酯(ps)、硫酸乙烯酯(dtd)中的至少一种;链状酯选自碳酸二甲酯(dmc)、碳酸丁烯酯、碳酸二乙酯(dec)、碳酸二丙酯、碳酸甲乙酯(emc)、碳酸甲丙酯、碳酸乙丙酯、甲酸甲酯、甲酸乙酯、甲酸丙酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、丙酸甲酯、丙酸乙酯、丙酸丙酯中的至少一种。
[0113]
相应地,本发明实施例还提供了一种二次电池,其包括上述的电解液。
[0114]
本发明实施例提供的二次电池,包括上述的电解液。该电解液可以有效改善所得二次电池的安全性和稳定性,经实验证明,本发明提供的二次电池在常温、高温下均具有良好的循环性能和存储性能,使用寿命更长。
[0115]
具体地,本发明实施例提供的二次电池包括正极、负极、电解液和隔膜,其中,正极包括正极集流体及其表面的正极活性材料层,用于制备正极活性材料层的正极活性浆料的组分包括正极活性物质、导电剂和粘结剂,正极活性物质选自li、na或k的过渡金属氧化物。在一些实施例中,正极活性物质选自licoo2、limn2o4、limno2、li2mno4、lifepo4、lini
x
coymnzo2、li
1 a
mn
1-xnx
o2、lico
1-xnx
o2、life
1-xnx
po4、limn
2-y
nyo4、li2mn
1-x
o4,其中,0≤a《0.2,0≤x,y,z≤1;n选自fe、ni、co、mn、zn、al、cr、mg、zr、mo、w、v、ti、b、f和y中的至少一种,且正极活性物质的质量占正极活性浆料的质量的88%-98%。可以理解的是,以上正极活性物质的具体选择是以li的过渡金属氧化物为例进行说明,根据实际需要,li可替换为na或k。
[0116]
负极包括负极集流体及其表面的负极活性材料层,用于制备负极活性材料层的负极活性浆料的组分包括负极活性物质、导电剂、粘结剂、增稠剂。在一些实施例中,负极活性物质选自天然石墨、人造石墨、软碳、硬碳、钛酸锂、硅、硅碳合金、硅氧合金等能够发生锂离子嵌入脱出反应的负极活性物质中的至少一种,且负极活性物质的质量占负极活性浆料的质量的90%-96%。
[0117]
需要说明的是,上述正极集流体(或负极集流体)和正极活性材料层(或负极活性
材料层)仅提供了一种常用的位置关系,即将正极活性浆料(或负极活性浆料)涂覆于正极集流体(或负极集流体)表面形成正极活性材料层(或负极活性材料层),不应理解为其是对本发明实施例提供的二次电池的限制。根据实际情况,结合对电池性能的要求可对集流体和活性材料进行改变,如将正极活性物质(或负极活性物质)及助剂的混合粉料填充在空心正极集流体(或空心负极集流体)的内部等各种方式。
[0118]
进一步地,制备正极活性浆料和负极活性浆料时还需要加入溶剂,该溶剂为高纯去离子水或n-甲基吡咯烷酮(nmp),其中,高纯度去离子水的电导率≤3us/cm,n-甲基吡咯烷酮的水分含量≤100ppm。
[0119]
进一步地,正、负极导电剂选自导电石墨、乙炔黑、纳米银粉中的至少一种,且正、负极导电剂的质量分别占正、负极活性浆料的质量的1%-6%。
[0120]
进一步地,正、负极粘结剂选自聚四氟乙烯、聚偏氟乙烯、聚偏氟乙烯-六氟丙烯、丙烯酸、丁苯橡胶中的至少一种,且正、负极粘结剂的质量分别占正、负极活性浆料的质量的1%-6%。
[0121]
进一步地,负极增稠剂为羧甲基纤维素钠,其质量占负极活性浆料的质量的1%-4%。
[0122]
在一些实施例中,隔膜具体可采用三层复合膜,其厚度为12μm-36μm,孔隙率为30%-70%。
[0123]
为使本发明上述实施细节和操作能清楚地被本领域技术人员理解,以及本发明实施例双草酸磷酸盐的制备方法、双草酸磷酸盐衍生物及其制备方法、电解质盐的进步性能显著的体现,以下通过多个实施例来举例说明上述技术方案。为了能够平行对比使能够更为直观的比较产物的制备过程,在本发明的实施例中使用的是同一种自制的反应中间体草酸硅基酯。
[0124]
实施例1
[0125]
本实施例提供了不同卤代硅烷化合物用于制备反应中间体草酸硅基酯的制备方法,具体如下:
[0126]
1.当卤代硅烷化合物为三甲基氯硅烷时
[0127]
向250ml的两口瓶中加入9g草酸和50ml碳酸二甲酯,室温搅拌草酸不完全溶解,将溶有23g me3sicl的30ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,滴加过程中草酸逐渐溶解,全部滴加完成后不溶物完全溶解,溶液为无色透明状。将体系降至0℃后加入21g三乙胺,保持温度为0℃搅拌3h后静置沉降后常压过滤,将滤液常压蒸馏除去溶剂得到粘稠淡黄色液体,然后60℃减压蒸馏得到无色液体22.1g,产率为94%,静置一段时间无色液体凝固变为白色粉末状固体,得到草酸二(三甲基硅基)酯。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过gc-ms进行分析,分析结果显示gc-ms(esi)[c8h
18
o4si2]-234.11,证明所得白色粉末状固体为草酸二(三甲基硅基)酯。
[0128]
2.当卤代硅烷化合物为叔丁基二甲基氯硅烷时
[0129]
向250ml的两口瓶中加入9g草酸和50ml碳酸二甲酯,室温搅拌草酸不完全溶解,将溶有31g t
bume2sicl的30ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,滴加过程中草酸逐渐溶解,全部滴加完成后不溶物完全溶解,溶液为无色透明状。将体系降至0℃后加入
21g三乙胺,保持温度为0℃搅拌3h后静置沉降后常压过滤,将滤液常压蒸馏除去溶剂得到粘稠淡黄色液体,然后60℃减压蒸馏得到无色液体30.6g,产率为96%,静置一段时间无色液体凝固变为白色粉末状固体,得到草酸二(叔丁基二甲基硅基)酯。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过gc-ms进行分析,分析结果显示gc-ms(esi)[c
14h30
o4si2]-318.20,证明所得白色粉末状固体为草酸二(叔丁基二甲基硅基)酯。
[0130]
3.当卤代硅烷化合物为二甲胺基二甲基氯硅烷时
[0131]
向250ml的两口瓶中加入9g草酸和50ml碳酸二甲酯,室温搅拌草酸不完全溶解,将溶有30g(me2n)me2sicl的30ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,滴加过程中草酸逐渐溶解,全部滴加完成后不溶物完全溶解,溶液为无色透明状。将体系降至0℃后加入21g三乙胺,保持温度为0℃搅拌3h后静置沉降后常压过滤,将滤液常压蒸馏除去溶剂得到粘稠淡黄色液体,然后60℃减压蒸馏得到无色液体28.7g,产率为98%,静置一段时间无色液体凝固变为白色粉末状固体,得到草酸二(二甲胺基二甲基硅基)酯。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过gc-ms进行分析,分析结果显示gc-ms(esi)
[0132]
[c
10h24
n2o4si2]-292.46,证明所得白色粉末状固体为草酸二(二甲胺基二甲基硅基)酯。
[0133]
为降低成本,以下实施例均以草酸二(三甲基硅基)酯作为中间体草酸硅基酯进行实验。
[0134]
实施例2
[0135]
本实施例提供了一种二氟双草酸磷酸锂(lidodfp)的制备方法,具体如下:
[0136]
(11)向250ml的两口瓶中加入15.2g lipf6和50ml碳酸二甲酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二甲酯溶液缓慢加入两口瓶中,加入过程中有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0137]
(12)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入40ml碳酸二甲酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体23.2g,产率为92%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为lidodfp。通过卡氏水分测定仪和电位滴定仪测定水分为13ppm,酸度为25ppm,氯离子浓度为1ppm。
[0138]
实施例3
[0139]
本实施例提供了一种二氟双草酸磷酸锂(lidodfp)的制备方法,具体如下:
[0140]
(21)向250ml的两口瓶中加入15.2g lipf6和50ml碳酸甲乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸甲乙酯溶液缓慢加入两口瓶中,加入过程中有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体
放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0141]
(22)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入40ml碳酸甲乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体23.5g,产率为93%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为lidodfp。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为23ppm,氯离子浓度为1ppm。
[0142]
实施例4
[0143]
本实施例提供了一种二氟双草酸磷酸锂(lidodfp)的制备方法,具体如下:
[0144]
(31)向250ml的两口瓶中加入15.2g lipf6和50ml碳酸二乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二乙酯溶液缓慢加入两口瓶中,加入过程中有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0145]
(32)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入40ml碳酸二乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体23.7g,产率为94%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为lidodfp。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为22ppm,氯离子浓度为1ppm。
[0146]
实施例5
[0147]
本实施例提供了一种二氟双草酸磷酸钠(nadodfp)的制备方法,具体如下:
[0148]
(41)向250ml的两口瓶中加入16.8g napf6和50ml碳酸二甲酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二甲酯溶液缓慢加入两口瓶中,加入过程中固体逐渐溶解同时有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0149]
(42)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入50ml碳酸二甲酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体24.9g,产率为93%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为nadodfp。通过卡氏水分测定仪和电位滴定仪测定水分
为15ppm,酸度为24ppm,氯离子浓度为1ppm。
[0150]
实施例6
[0151]
本实施例提供了一种二氟双草酸磷酸钠(nadodfp)的制备方法,具体如下:
[0152]
(51)向250ml的两口瓶中加入16.8g napf6和50ml碳酸甲乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸甲乙酯溶液缓慢加入两口瓶中,加入过程中固体逐渐溶解同时有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0153]
(52)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入50ml碳酸甲乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体24.6g,产率为92%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为nadodfp。通过卡氏水分测定仪和电位滴定仪测定水分为13ppm,酸度为25ppm,氯离子浓度为1ppm。
[0154]
实施例7
[0155]
本实施例提供了一种二氟双草酸磷酸钠(nadodfp)的制备方法,具体如下:
[0156]
(61)向250ml的两口瓶中加入16.8g napf6和50ml碳酸二乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二乙酯溶液缓慢加入两口瓶中,加入过程中固体逐渐溶解同时有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0157]
(62)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入50ml碳酸二乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体25.1g,产率为94%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为nadodfp。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为25ppm,氯离子浓度为1ppm。
[0158]
实施例8
[0159]
本实施例提供了一种二氟双草酸磷酸钾(kdodfp)的制备方法,具体如下:
[0160]
(71)向250ml的两口瓶中加入18.4g kpf6和50ml碳酸二甲酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二甲酯溶液缓慢加入两口瓶中,加入过程中固体逐渐溶解同时有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0161]
(72)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压
过滤向白色固体中加入50ml碳酸二甲酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体26.4g,产率为93%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为kdodfp。通过卡氏水分测定仪和电位滴定仪测定水分为10ppm,酸度为22ppm,氯离子浓度为1ppm。
[0162]
实施例9
[0163]
本实施例提供了一种二氟双草酸磷酸钾(kdodfp)的制备方法,包具体如下:
[0164]
(81)向250ml的两口瓶中加入18.4g kpf6和50ml碳酸甲乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸甲乙酯溶液缓慢加入两口瓶中,加入过程中有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0165]
(82)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入50ml碳酸甲乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体26.7g,产率为94%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为kdodfp。通过卡氏水分测定仪和电位滴定仪测定水分为13ppm,酸度为21ppm,氯离子浓度为1ppm。
[0166]
实施例10
[0167]
本实施例提供了一种二氟双草酸磷酸钾(kdodfp)的制备方法,具体如下:
[0168]
(91)向250ml的两口瓶中加入18.4g kpf6和50ml碳酸二乙酯,室温搅拌固体完全溶解,然后将溶有46g实施例1所得草酸硅基酯的50ml碳酸二乙酯溶液缓慢加入两口瓶中,加入过程中固体逐渐溶解同时有me3sif气体放出。全部加完后室温继续搅拌3h然后升温至40℃,有大量气体放出,保持温度为40℃反应1h。溶液为无色透明状,反应产生的气体通过氢氧化钠饱和水溶液吸收。
[0169]
(92)将体系降至室温静置沉降后常压过滤,将滤液浓缩析出大量白色固体。常压过滤向白色固体中加入50ml碳酸二乙酯,升温至60℃使白色固体完全溶解,然后降至室温浓缩结晶,析出大量的白色晶体,将白色晶体置于40℃烘箱真空干燥3h得到白色粉末状固体26.4g,产率为93%。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)c4o8pf2[dodfp]-245.02,证明所得白色粉末状固体为kdodfp。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为21ppm,氯离子浓度为1ppm。
[0170]
实施例11
[0171]
本实施例提供了一种双草酸氰基氟磷酸锂的制备方法,具体如下:
[0172]
向250ml双口瓶中加入实施例4所得lidodfp 25.2g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将10g me3sicn(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸氰基氟磷酸锂23.8g,产率为92%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfcn]-252.08,证明所得白色粉末状固体为双草酸氰基氟磷酸锂。通过卡氏水分测定仪和电位滴定仪测定水分为10ppm,酸度为22ppm,氯离子浓度为0.5ppm。
[0173]
实施例12
[0174]
本实施例提供了一种双草酸氰基氟磷酸钠的制备方法,具体如下:
[0175]
向250ml双口瓶中加入实施例7所得nadodfp 26.8g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将10g me3sicn(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸氰基氟磷酸钠25.3g,产率为92%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfcn]-252.08,证明所得白色粉末状固体为双草酸氰基氟磷酸钠。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为20ppm,氯离子浓度为0.5ppm。
[0176]
实施例13
[0177]
本实施例提供了一种双草酸氰基氟磷酸钾的制备方法,具体如下:
[0178]
向250ml双口瓶中加入实施例9所得kdodfp 28.4g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将10g me3sicn(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸氰基氟磷酸钾26.8g,产率为92%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfcn]-252.08,证明所得白色粉末状固体为双草酸氰基氟磷酸钾。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为18ppm,氯离子浓度为0.5ppm。
[0179]
实施例14
[0180]
本实施例提供了一种双草酸二氰基磷酸锂的制备方法,具体如下:
[0181]
向250ml双口瓶中加入实施例4所得lidodfp 25.2g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将20g me3sicn(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二氰基磷酸锂25.0g,产率为94%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pc2n2]-259.06,证明所得白色粉末状固体为双草酸二氰基磷酸锂。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为19ppm,氯离子浓度为0.5ppm。
[0182]
实施例15
[0183]
本实施例提供了一种双草酸二氰基磷酸钠的制备方法,具体如下:
[0184]
向250ml双口瓶中加入实施例7所得nadodfp 26.8g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将20g me3sicn(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二氰基磷酸钠26.5g,产率为94%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pc2n2]-259.06,证明所得白色粉末状固体为双草酸二氰基磷酸钠。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为16ppm,氯离子浓度为0.5ppm。
[0185]
实施例16
[0186]
本实施例提供了一种双草酸二氰基磷酸钾的制备方法,具体如下:
[0187]
向250ml双口瓶中加入实施例9所得kdodfp 28.4g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将20g me3sicn(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二氰基磷酸钾28.0g,产率为94%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pc2n2]-259.06,证明所得白色粉末状固体为双草酸二氰基磷酸钾。通过卡氏水分测定仪和电位滴定仪测定水分为9ppm,酸度为15ppm,氯离子浓度为0.5ppm。
[0188]
实施例17
[0189]
本实施例提供了一种双草酸异氰酸酯氟磷酸锂的制备方法,具体如下:
[0190]
向250ml双口瓶中加入实施例4所得lidodfp 25.2g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将11.5g me3sinco(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸异氰酸酯氟磷酸锂25.6g,产率为93%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfnco]-268.04,证明所得白色粉末状固体为双草酸异氰酸酯氟磷酸锂。通过卡氏水分测定仪和电位滴定仪测定水分为8ppm,酸度为12ppm,氯离子浓度为0.5ppm。
[0191]
实施例18
[0192]
本实施例提供了一种双草酸异氰酸酯氟磷酸钠的制备方法,具体如下:
[0193]
向250ml双口瓶中加入实施例7所得nadodfp 26.8g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将11.5g me3sinco(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸异氰酸酯氟磷酸钠27.1g,产率为93%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfnco]-268.04,证明所得白色粉末状固体为双草酸异氰酸酯氟磷酸钠。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为20ppm,氯离子浓度为0.5ppm。
[0194]
实施例19
[0195]
本实施例提供了一种双草酸异氰酸酯氟磷酸钾的制备方法,具体如下:
[0196]
向250ml双口瓶中加入实施例9所得kdodfp 26.8g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将11.5g me3sinco(0.1mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸异氰酸酯氟磷酸钾28.6g,产率为93%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pfnco]-268.04,证明所得白色粉末状固体为双草酸异氰酸酯氟磷酸钾。通过卡氏水分测定仪和电位滴定仪测定水分为11ppm,酸度为14ppm,氯离子浓度为0.5ppm。
[0197]
实施例20
[0198]
本实施例提供了一种双草酸二异氰酸酯磷酸锂的制备方法,具体如下:
[0199]
向250ml双口瓶中加入实施例4所得lidodfp 25.2g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将23g me3sinco(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二异氰酸酯磷酸锂28.0g,产率为94%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pn2c2o2]-291.03,证明所得白色粉末状固体为双草酸二异氰酸酯磷酸锂。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为16ppm,氯离子浓度为0.5ppm。
[0200]
实施例21
[0201]
本实施例提供了一种双草酸二异氰酸酯磷酸钠的制备方法,具体如下:
[0202]
向250ml双口瓶中加入实施例7所得nadodfp 26.8g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将23g me3sinco(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二异氰酸酯磷酸钠29.5g,产率为94%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pn2c2o2]-291.03,证明所得白色粉末状固体为双草酸二异氰酸酯磷酸钠。通过卡氏水分测定仪和电位滴定仪测定水分为12ppm,酸度为18ppm,氯离子浓度为0.5ppm。
[0203]
实施例22
[0204]
本实施例提供了一种双草酸二异氰酸酯磷酸钾的制备方法,具体如下:
[0205]
向250ml双口瓶中加入实施例9所得kdodfp 28.4g(0.1mol)和50ml碳酸二甲酯,室温搅拌固体完全溶解在溶剂中,将23g me3sinco(0.2mol)的50ml碳酸二甲酯溶液通过滴液漏斗加入到两口瓶中,控制滴加速度为1滴/秒,滴加过程中有气体放出并且溶液为无色透明状,全部滴加完成后室温继续搅拌3h然后升温至40℃反应1h。室温静置常压过滤除去悬浮固体杂质,得到无色透明溶液,室温减压浓缩得到白色固体,将白色固体使用40ml碳酸二甲酯重结晶然后60℃真空干燥3h得到目标产物双草酸二异氰酸酯磷酸钾30.7g,产率为93%。反应产生的气体通过氢氧化钠饱和水溶液吸收。在手套箱中,取所得白色粉末状固体5mg,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过液质联用(thermo fisher scientific)进行分析,分析结果显示lc-ms(esi)[c4o8pn2c2o2]-291.03,证明所得白色粉末状固体为双草酸二异氰酸酯磷酸钾。通过卡氏水分测定仪和电位滴定仪测定水分为10ppm,酸度为13ppm,氯离子浓度为0.5ppm。
[0206]
实验例1
[0207]
将六氟磷酸锂(lipf6)溶于碳酸酯溶剂中,使其浓度为1.0mol/l,所用碳酸酯溶剂
为碳酸乙烯酯、碳酸甲乙酯和碳酸二乙酯按照质量比为3:5:2的混合溶剂,且质量分数为70%-90%,同时加入质量分数为1%的碳酸亚乙烯酯(vc)、质量分数为1%的1,3-丙磺酸内酯(ps)和质量分数为1%的硫酸乙烯酯(dtd),得到对照样品电解液,编号记为(1);
[0208]
在氩气氛围的手套箱中(手套箱中水、氧含量低于1ppm)分别将六氟磷酸锂与实施例4所得lidodfp、实施例11所得双草酸氰基氟磷酸锂、实施例14所得双草酸二氰基磷酸锂、实施例17所得双草酸异氰酸酯氟磷酸锂、实施例20所得双草酸二异氰酸酯磷酸锂以等摩尔比(均为0.5mol/l)配制电解液,同时加入质量分数为1%的碳酸亚乙烯酯、质量分数为1%的1,3-丙磺酸内酯和质量分数为1%的硫酸乙烯酯。分别得到编号为(2)-(6)的电解液,编号(1)-(6)的电解液锂离子浓度均为1.0mol/l。
[0209]
在氩气氛围的手套箱中(手套箱中水、氧含量低于1ppm)分别将实施例4所得lidodfp、实施例11所得双草酸氰基氟磷酸锂、实施例14所得双草酸二氰基磷酸锂、实施例17所得双草酸异氰酸酯氟磷酸锂、实施例20所得双草酸二异氰酸酯磷酸锂溶于碳酸酯溶剂中,使其浓度为1.0mol/l,所用碳酸酯溶剂为碳酸乙烯酯、碳酸甲乙酯和碳酸二乙酯按照质量比为3:5:2的混合溶剂,同时加入质量分数为1%的碳酸亚乙烯酯、质量分数为1%的1,3-丙磺酸内酯和质量分数为1%的硫酸乙烯酯,分别得到编号为(7)-(11)的电解液。
[0210]
将制备所得电解液(1)-(11)密封并储存在手套箱中,分别取少量电解液放置于氟化瓶中,室温放置1个月后,通过卡尔费休水分仪测试其水分,通过酸碱滴定法测试其酸度。具体数据如表1所示。
[0211]
表1电解液水分和酸度测试结果
[0212]
电解液编号电解质盐水分(ppm)酸度(ppm)(1)lipf680125(2)lipf6 lidodfp76111(3)lipf6 双草酸氰基氟磷酸锂72117(4)lipf6 双草酸二氰基磷酸锂5284(5)lipf6 双草酸异氰酸酯氟磷酸锂6882(6)lipf6 双草酸二异氰酸酯磷酸锂4871(7)lidodfp71105(8)双草酸氰基氟磷酸锂63110(9)双草酸二氰基磷酸锂4275(10)双草酸异氰酸酯氟磷酸锂5974(11)双草酸二异氰酸酯磷酸锂4163
[0213]
表1的结果表明,使用本发明实施例所得的电解质盐作为主盐与六氟磷酸锂组合使用,或单独使用,均有利于降低电解液的水分和酸度。
[0214]
实验例2
[0215]
本实验例制作了软包锂离子电池,包括正极材料、负极材料、陶瓷涂覆的pe隔膜材料、铝塑膜以及实验例1所得编号为(1)-(11)的电解液。
[0216]
其中,以镍钴锰酸锂(lini
0.6
co
0.2
mn
0.2
o2,简称ncm622)三元材料作为正极活性物质,人造石墨作为负极活性物质,正极活性浆料按质量比96%正极活性物质 2%pvdf粘合剂 2%super p导电炭黑溶于溶剂n-甲基吡咯烷酮中混合得到。然后将正极活性浆料均匀
涂布在集流体铝箔上,涂布量为280g/m2,随后在80℃下烘干后进行冷压、切边、裁片、分条后,在80℃真空条件下干燥4h,焊接极耳,得到正极片。负极活性浆料按质量比96%负极活性物质 2%cmc/sbr粘合剂 2%super p导电炭黑混合后加入去离子水中搅拌均匀得到,然后将负极活性浆料均匀涂布在集流体铜箔上,涂布量为200g/m2,随后在85℃下烘干后进行冷压、切边、裁片、分条后,在110℃真空条件下干燥4h,焊接极耳,得到负极片。将正极片、负极片以及陶瓷涂覆的pe隔膜经过叠片工艺制作成小软包电芯,并在75℃下真空烘烤10h,分别使用实验例1所得编号为(1)-(13)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,分别得到软包电池依次为(a)-(k)。
[0217]
为了保持实验的一致性,所有软包电池都使用相同体积的电解液。然后对制备好的电池进行充放电测试,使用land充放电测试系统对组装好的电池进行电化学性能测试。电池测试电压为3.0-4.2v,分别在室温和45℃条件下以0.33c恒流充放电循环300周后测试电池的容量保持率、分别在室温下储存1个月和在60℃下存储15天后的容量保持率,以及在60℃下存储30天后的电芯体积膨胀率,具体数据如表2所示。
[0218]
表2电池性能测试结果
[0219][0220][0221]
通过表2的测试结果可知,使用本发明实施例所得电解质盐作为主盐与六氟磷酸锂组合使用,或单独使用,均有利于提升电池的常温循环、高温循环性能、电池常温存储性能和高温存储性能,同时可以抑制电池产气,减小电池的体积膨胀,提高电池安全性和使用寿命。
[0222]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
