1.本发明属于医药技术领域,具体涉及一种右兰索拉唑的制备方法。
背景技术:
2.右兰索拉唑(dexlansoprazole)是日本武田制药公司研发的食管炎治疗新药,2009年1月30日经美国fda批准上市,该药是质子泵抑制剂兰索拉唑的单对映体,用于治疗与非糜烂性胃食管反流病相关的胃灼热及不同程度糜烂性食道炎,比兰索拉唑具有更高的生物利用度和更少副作用。其结构式如式(i)所示:
[0003][0004]
cn1117747c公开了光学纯兰索拉唑的制备方法,以光学纯的联二萘酚类化合物为手性助剂,采用拆分方法制得粗品,再在碱性溶液或水/少量有机溶剂中制备成较稳定的白色粉末状的光学纯兰索拉唑,这种手性拆分法原料利用率低,成本较高。
[0005]
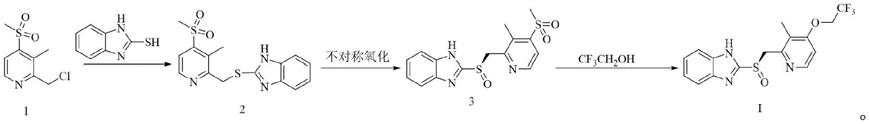
wo2010035283a2公开了一种氯甲基化合物1与苯并巯基咪唑缩合,得到硫醚2,硫醚经过氧化氢异丙苯氧化得到化合物3,最后与三氟乙醇在碱叔丁醇钾作用下生成右兰索拉唑的制备方法。该方法在最后一步合成时,三氟乙醇可能与甲磺酰基反应,促使消耗更多的三氟乙醇:
[0006][0007]
cn106543146a公开了一种以化合物4为起始物料,经氯代、亲核取代,不对称氧化三步反应制备右兰索拉唑:
[0008][0009]
cn102108077b公开了在氢氧化钠条件下,以r-2-(((4-氯-3-甲基-2-吡啶基)甲基)亚磺酰基)苯并咪唑为原料,经不对称氧化及三氟乙醇取代制得右兰索拉唑。但是,反应过程中会生成水,而水的存在对于反应的进行是极为不利的,因此会导致原料反应不完全,砜型杂质较大,给后续精制带来很大压力,需要通过柱层析方法来除去砜型类似物等杂质,延长了工艺周期,增加了生产成本;
[0010][0011]
右兰索拉唑的手性构建为关键步骤,主要方法有手性拆分,不对称氧化等,经过文献对比发现,目前应用较多的是不对称氧化方法,但是,不对称氧化的过程中,若反应条件控制不当,则会产生砜型杂质a和n-氧化物b等杂质,这些杂质难以通过重结晶等常规的纯化方法除去,通常需要通过色谱法除杂,该除杂方法成本过高;
[0012][0013]
综上,为了减少工艺中有关物质并同时优化产率,研究人员进一步研究了对映选择性氧化方法。在保证产物收率与纯度的基础上,研究一种制备成本较低,且环境友好的右兰索拉唑的制备工艺。
技术实现要素:
[0014]
本发明所要解决的技术问题是克服现有技术的不足,提供一种可控的不对称氧化方法,以salen络合物催化体系作为手性催化剂,以次氯酸钠作为氧化剂,该反应体系,反应条件更温和,过度氧化杂质含量明显降低,降低了后处理的压力。
[0015]
本发明的方法包括以下步骤:
[0016][0017]
步骤一:化合物ii在手性催化剂作用下,与次氯酸钠发生氧化反应生成化合物i粗品,
[0018]
步骤二:化合物i粗品经有机溶剂重结晶得手性纯度高的化合物i。
[0019]
作为一种具体的实施方式,步骤一中所述手性催化剂选自化合物iii或iv,优选化合物iv。
[0020]
[0021]
作为一种具体的实施方式,步骤一中反应溶剂选自乙酸乙酯、乙腈、丙酮中的一种,优选乙腈。
[0022]
作为一种具体的实施方式,步骤一中化合物ii与次氯酸钠投料摩尔比为1:1-1:2,优选1:1.2;所述催化剂摩尔用量为化合物ii的0.05~0.2倍。
[0023]
作为一种具体的实施方式,步骤一中的反应温度为0~30℃,优选15~25℃。
[0024]
作为一种具体的实施方式,步骤二中所述重结晶溶剂选自四氢呋喃、甲醇、乙醇、乙酸乙酯中的一种与水的混合溶液,优选四氢呋喃与水的混合溶剂。
[0025]
本发明的有益效果:使用价廉,环境友好,安全性高的次氯酸钠作为氧化剂,代替传统的过氧化氢异丙苯、间氯过氧苯甲酸等氧化剂,减少了过度氧化反应,降低后处理压力。使用新的催化剂体系,代替条件敏感的l-( )-酒石酸二乙酯/异丙醇钛催化体系,该催化剂体系反应条件温和,催化效率高。由于有效控制了副反应,降低了后处理及精制压力,使得总收率相对文献方法由33.3%提升至76%。
具体实施方式
[0026]
下面结合具体实施例对本发明作进一步的详细说明。以下实施例用于理解本发明的方法和核心思想,对于本领域的技术人员来说,在不脱离本发明构思的前提下,进行任何可能的变化或替换,均属于本发明的保护范围。本发明实施例中未注明具体条件的实验方法,通常为常规条件,或按照原料或商品制造厂商所建议的条件;未注明来源的试剂,通常为通过商业途径可购得的常规试剂。
[0027]
本技术实施例所使用的术语“手性配体”是指催化剂iii和iv在未结合锰时的手性结构,可以通过常规的商业途径获得;更进一步地,本技术催化剂iii和iv分别对应的手性配体的结构如iiia和ivb所示:
[0028][0029]
实施例1
[0030]
催化剂iii/iv的制备:手性配体(0.5mol)溶解于无水乙醇中,并向其中加入溶有mn(oac)4的无水乙醇溶液,回流反应4小时。将反应液降至室温,然后加入1.5mol氯化锂,25℃搅拌2小时,过滤得产品。依次用乙醇,蒸馏水洗涤数次,干燥得固体,备用。
[0031]
次氯酸钠溶液配制:取次氯酸钠100g溶于水,加入300ml水搅拌溶解,调节体系ph至11~12,备用。
[0032]
步骤一:取化合物ii(35.3g,0.1mol),催化剂iv(9.51g,0.015mol)及乙腈220g搅拌,控温20℃,加入预先配好的次氯酸钠溶液(8.2g,0.11mol),搅拌反应2-3小时,反应结束后,使用冰醋酸溶液调节体系ph约为9.0,滤除催化剂后,然后加入纯化水220ml,降温至5℃,搅拌析晶3小时,抽滤,得粗品35g,纯度97.2%,ee值:98.5%,氧化杂质a:0.4%,氧化杂质b:0.5%。
[0033]
步骤二:取右兰索拉唑粗品35g,加入四氢呋喃350ml,搅拌升温至45℃,保温搅拌30分钟至体系完全溶解,加入活性炭脱色,热过滤,滤液缓慢降温至20℃,滴加纯化水525g,加完后,降温至5℃,析晶2小时,过滤,干燥得纯品i30g,收率85.7%,纯度99.2%,ee值:99.5%。两步收率约76%。
[0034]
实施例2
[0035]
步骤一:取化合物ii(35.3g,0.1mol),催化剂iii(9.69g,0.015mol)及乙腈220g搅拌,控温20℃,加入预先配好的次氯酸钠溶液(8.2g,0.11mol),搅拌反应2-3小时,反应结束后,使用冰醋酸溶液调节体系ph约为9.0,滤除催化剂后,然后加入纯化水220ml,降温至5℃,搅拌析晶3小时,抽滤,得粗品34.8g,纯度96.8%,ee值:98.8%,氧化杂质a:0.5%,氧化杂质b:0.5%。
[0036]
步骤二:取右兰索拉唑粗品34g,加入四氢呋喃340ml,搅拌升温至45℃,保温搅拌30分钟至体系完全溶解,加入活性炭脱色,热过滤,滤液缓慢降温至20℃,滴加纯化水525g,加完后,降温至5℃,析晶2小时,过滤,干燥得纯品i30g,收率88.2%,纯度99.1%,ee值:99.4%。
[0037]
实施例3
[0038]
步骤一:取化合物ii(35.3g,0.1mol),催化剂iv(9.51g,0.015mol)及乙腈220g搅拌,控温20℃,加入预先配好的次氯酸钠溶液(8.2g,0.11mol),搅拌反应2-3小时,反应结束后,使用冰醋酸溶液调节体系ph约为9.0,滤除催化剂后,然后加入纯化水220ml,降温至5℃,搅拌析晶3小时,抽滤,得粗品35g,纯度97.2%,ee值:98.5%,氧化杂质a:0.4%,氧化杂质b:0.5%。
[0039]
步骤二:取右兰索拉唑粗品35g,加入甲醇350ml,搅拌升温至55℃,保温搅拌30分钟至体系完全溶解,加入活性炭脱色,热过滤,滤液缓慢降温至20℃,滴加纯化水525g,加完后,降温至5℃,析晶2小时,过滤,干燥得纯品i28g,收率80%,纯度99.5%,ee值:99.5%。
[0040]
实施例4对比实施例-采用cn106866630b方法(其报道的实施例1[0043]-[0051]中经历复杂的后处理过程,两步总收率为32.3%)
[0041]
步骤一:取化合物ii(35.3g,0.1mol),甲苯176ml至反应瓶中,升温至回流并保持约60分钟,在氮气保护下冷却反应物料至室温,加水0.3g和l-( )-酒石酸二乙酯15.8g,升温至60~65℃,保温约15分钟后于10~15分钟内加入异丙醇钛10.2g,保温反应约50分钟后冷却至室温,再加入二异丙基乙胺8.4g,然后在约30分钟内加入氢过氧化枯烯21.8g,保持反应约3.5小时。反应结束后,使用硫代硫酸钠溶液洗涤三次,除去水层。升温至30~35℃,加水45g,然后在10~15分钟内加入甲基叔丁基醚89ml,接着在10~15分钟内加入环己烷177ml,搅拌不超过40分钟。过滤,用甲基叔丁基醚35ml洗涤,干燥约1.5小时。滤饼中加入丙酮177ml,搅拌约10分钟后于30~45分钟内投入530ml水,搅拌约30分钟后,过滤,干燥右兰索拉唑粗品30.1g,收率81.5%。
[0042]
室温下,右兰索拉唑粗品30g,丙酮150ml至反应瓶,搅拌,加氨水溶液0.7ml,活性炭1.73g。过滤,用丙酮淋洗,收集滤液至反应瓶中。加氨水溶液0.7ml,过滤,滤饼中再加入二氯甲烷300ml,搅拌后静置,分离底部有机层,用硫代硫酸钠干燥。蒸馏除去有机溶剂,再加入丙酮30ml,蒸馏除去溶剂,冷却至室温后加丙酮30ml,搅拌使完全溶解。投入正庚烷300ml,升温至40~45℃后过滤、静置约30分钟。过滤,用正庚烷洗涤,干燥右兰索拉唑10g,
收率33.3%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。