1.本发明属于医药技术领域,特别是一种乌蛇止痒丸的指纹图谱构建和检测方法。
背景技术:
2.乌蛇止痒丸由乌梢蛇、防风、蛇床子、关黄柏、苍术、红参须、牡丹皮、蛇胆汁、苦参、人工牛黄和当归等11味中药组成,具有养血祛风、燥湿止痒的功效,主要用于风湿热邪蕴于肌肤所致的慢性荨麻疹、皮肤瘙痒症。作为一种中成药,乌蛇止痒丸成分复杂,一般通过多靶点、多作用途径起效。因此,基于多成分对乌蛇止痒丸进行质量控制尤为重要。
3.而传统的乌蛇止痒丸质量控制方法主要是基于单个成分进行的,例如已报道的乌蛇止痒丸的薄层色谱鉴别、盐酸小檗碱的hplc含量测定方法(王蔚等发表的“rp
‑
hplc法测定乌蛇止痒丸中小檗碱含量”以及2020版《中国药典》)以及苦参碱的薄层扫描含量测定方法(郑晓晴等发表的“乌蛇止痒丸质量标准的研究”)。
4.基于单个成分对乌蛇止痒丸进行质量检测不能全面反映乌蛇止痒丸的物质基础,进而无法实现对乌蛇止痒丸的整体质量控制及评价。因此,如何全面反映乌蛇止痒丸的物质基础从而为乌蛇止痒丸的整体质量控制和评价提供有效技术手段是目前亟待解决的技术问题。
技术实现要素:
5.基于以上背景技术,本发明的主要目的是提供一种乌蛇止痒丸的指纹图谱构建方法,采用该方法构建的指纹图谱结合多种化学成分的定量分析能够全面地反映乌蛇止痒丸整体化学成分,为乌蛇止痒丸的整体质量控制及评价提供了有效手段。本发明的另一目的是提供一种乌蛇止痒丸的检测方法。本发明的还一目的是提供一种乌蛇止痒丸的多成分含量检测方法。
6.本发明的目的可以通过以下技术方案实现:
7.一种乌蛇止痒丸的指纹图谱的构建方法,所述构建方法包括如下步骤:
8.取对照品和乌蛇止痒丸样品,分别制备对照品溶液和供试品溶液;采用高效液相色谱法对所述对照品溶液和所述供试品溶液进行检测,获得相应的检测图谱和对照图谱;将所述检测图谱导入中药色谱指纹图谱相似度评价系统进行分析,生成标准指纹图谱,参照所述对照图谱对所述标准指纹图谱的色谱峰进行指认,获得乌蛇止痒丸的指纹图谱;
9.所述对照品包含人参皂苷rg1、人参皂苷re、人参皂苷rb1、人参皂苷rb2、人参皂苷rb3、人参皂苷rc、人参皂苷f1、人参皂苷rd、人参皂苷f2、人参皂苷rg2、人参皂苷rg3、5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、亥茅酚苷、蛇床子素、欧前胡素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸黄柏碱、盐酸药根碱、丹皮酚、芍药苷、丹皮酚原苷、苦参碱、氧化苦参碱、槐果碱、氧化槐果碱、木兰花碱、掌叶防已碱、柠檬苦素、黄柏酮、胆红素和二氢欧山芹醇当归酸酯中的至少一种;
10.检测所采用条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲
酸水溶液,流动相b为乙腈,采用梯度洗脱。
11.在其中一个实施例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比为5%;5min~35min,所述流动相b的体积百分比由5%升高至20%;35min~40min,所述流动相b的体积百分比为20%;40min~48min,所述流动相b的体积百分比由20%升高至23%;48min~60min,所述流动相b的体积百分比由23%升高至35%;60min~90min,所述流动相b的体积百分比由35%升高至55%;90min~105min,所述流动相b的体积百分比由55%升高至95%;105min~115min,所述流动相b的体积百分比为95%。
12.在其中一个实施例中,所述固定相中填料的粒径为1.5μm~3μm。
13.在其中一个实施例中,检测所采用条件还包括:波长为230nm~300nm。
14.在其中一个实施例中,检测所采用条件还包括:流速为0.1ml/min~0.6ml/min。
15.在其中一个实施例中,检测所采用条件还包括:柱温为20℃~50℃。
16.在其中一个实施例中,检测所采用条件还包括:进样量为1μl~6μl。
17.在其中一个实施例中,制备所述对照品溶液采用的溶解溶剂含有甲醇。
18.在其中一个实施例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述乌蛇止痒丸样品进行提取,收集提取液。
19.在其中一个实施例中,所述提取溶剂含甲醇的体积百分比为10%~100%。
20.在其中一个实施例中,提取的方式采用超声提取。
21.在其中一个实施例中,收集所述提取液的方式采用离心或者过滤。
22.在其中一个实施例中,所述特征图谱包含34个特征峰,其中,7号峰为升麻素苷,9号峰为升麻素、14号峰为5
‑
o
‑
甲基维斯阿米醇苷,15号峰为药根碱,20号峰为小檗碱,21号峰为巴马汀,22号峰为丹皮酚,23号峰为佛手柑内酯,25号峰为花椒毒素,30号峰为花椒毒酚,32号峰为蛇床子素。
23.一种乌蛇止痒丸的检测方法,所述检测方法包括如下步骤:
24.取待测样品,制备供试品溶液,采用高效液相色谱法对所述供试品溶液进行检测,并将所得检测图谱与如上构建获得的指纹图谱进行比较;
25.检测所采用条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲酸水溶液,流动相b为乙腈,采用梯度洗脱。
26.在其中一个实施例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比为5%;5min~35min,所述流动相b的体积百分比由5%升高至20%;35min~40min,所述流动相b的体积百分比为20%;40min~48min,所述流动相b的体积百分比由20%升高至23%;48min~60min,所述流动相b的体积百分比由23%升高至35%;60min~90min,所述流动相b的体积百分比由35%升高至55%;90min~105min,所述流动相b的体积百分比由55%升高至95%;105min~115min,所述保持流动相b的体积百分比为95%。
27.在其中一个实施例中,所述固定相中填料的粒径为1.5μm~3μm。
28.在其中一个实施例中,检测所采用条件还包括:波长为230nm~300nm。
29.在其中一个实施例中,检测所采用条件还包括:流速为0.1ml/min~0.6ml/min。
30.在其中一个实施例中,检测所采用条件还包括:柱温为20℃~50℃。
31.在其中一个实施例中,检测所采用条件还包括:进样量为1μl~6μl。
32.在其中一个实施例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述待测
样品进行提取,收集提取液。
33.在其中一个实施例中,所述提取溶剂含甲醇的体积百分比为10%~100%。
34.在其中一个实施例中,提取的方式采用超声提取。
35.在其中一个实施例中,收集所述提取液的方式采用离心或者过滤。
36.一种乌蛇止痒丸的多成分含量检测方法,所述检测方法包括如下步骤:
37.取对照品和待测样品,分别制备对照品溶液和供试品溶液;采用高效液相色谱质谱联用法对所述对照品溶液和所述供试品溶液进行检测,并根据检测所述对照品溶液所得图谱和检测所述供试品溶液所得图谱中各特征峰的峰面积,确定所述乌蛇止痒丸中对应成分的含量;
38.所述对照品包含苦参碱、槐果碱、氧化苦参碱、升麻素、盐酸黄柏碱、木兰花碱、升麻素苷、盐酸小檗碱、盐酸巴马汀、丹皮酚、花椒毒素、佛手柑内酯、人参皂苷rb1、人参皂苷rb2、人参皂苷rc和二氢欧山芹醇当归酸酯中的至少一种;
39.所述高效液相色谱质谱联用法中,液相色谱条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲酸水溶液,流动相b为乙腈,采用梯度洗脱。
40.在其中一个实施例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比由5%升高至25%;5min~7min,所述流动相b的体积百分比由25%升高至37%;7min~8min,所述流动相b的体积百分比为37%;8min~10min,所述流动相b的体积百分比由37%升高至55%;10min~12min,所述流动相b的体积百分比由55%升高至95%;12min~14min,所述流动相b的体积百分比为95%;14min~15min,所述流动相b的体积百分比由95%下降至5%;15min~17min,所述流动相b的体积百分比为5%。
41.在其中一个实施例中,所述固定相中填料的粒径为1.5μm~3μm。
42.在其中一个实施例中,所述液相色谱条件还包括:流速为0.1ml/min~0.6ml/min。
43.在其中一个实施例中,所述液相色谱条件还包括:柱温为20℃~50℃。
44.在其中一个实施例中,所述液相色谱条件还包括:进样量为1μl~6μl在其中一个实施例中,所述高效液相色谱质谱联用法中,质谱条件包括:esi正离子模式,毛细管电压:2.5kv~3.5kv,源温度:145℃~155℃,脱溶剂温度:335℃~345℃,脱溶剂气流:640l/h~660l/h,锥形气流:25l/h~35l/h,碰撞气体:氩,脱溶剂气体:氮气。在其中一个实施例中,制备所述对照品溶液采用的溶解溶剂含有甲醇。
45.在其中一个实施例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述待测样品进行提取,收集提取液。
46.在其中一个实施例中,所述提取溶剂含甲醇的体积百分比为10%~100%。
47.在其中一个实施例中,提取的方式采用超声提取。
48.在其中一个实施例中,收集所述提取液的方式采用离心或者过滤。
49.与现有技术相比较,本发明具有如下有益效果:
50.本发明选用合适的对照品并搭配合适的高效液相色谱条件,构建得到乌蛇止痒丸的指纹图谱,该指纹图谱色谱峰丰富,且同时包含防风、蛇床子、关黄柏、牡丹皮、当归等多味药材的有效成分,能够更全面地反应乌蛇止痒丸的整体化学成分,提高了乌蛇止痒丸质量控制的准确性和全面性,为乌蛇止痒丸的整体质量控制及评价提供了有效手段。
51.本发明提供的乌蛇止痒丸的多成分含量检测方法,能够在短时间内测定乌蛇止痒
丸中的多种化学成分,是一种快速的乌蛇止痒丸的质量控制方法。
附图说明
52.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
53.图1为16个批次的乌蛇止痒丸样品的uplc图谱;
54.图2为乌蛇止痒丸标准指纹图谱;
55.图3为采用不同提取溶剂所得样品色谱图;
56.图4为不同提取时间条件下所得样品色谱图;
57.图5为采用不同色谱柱分离所得色图谱;
58.图6为不同流动相体系分离所得色谱图;
59.图7为不同流动相添加剂分离所得色谱图;
60.图8为不同色谱柱温下分离所得色谱图;
61.图9为不同检测器样品检测色谱图;
62.图10为梯度洗脱样品色谱图;
63.图11为非最优梯度洗脱条件下的样品色谱图;
64.图12为检测供试品y04006m所得检测图谱与指纹图谱比较图;
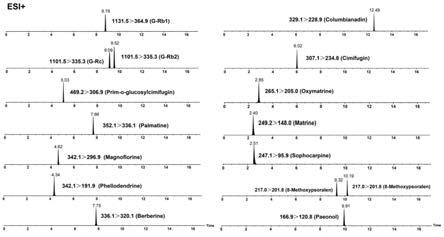
65.图13为乌蛇止痒丸样品中16种目标组分的代表性mrm色谱图;
66.图14为g
‑
rc(人参皂苷rc)和g
‑
rb2(人参皂苷rb2)使用三种不同色谱柱的分离性能比较图;
67.图15为16种对照品在正离子模式下的mrm色谱图。
具体实施方式
68.为了便于理解本发明,下面将对本发明进行更详细的描述。但是,应当理解,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式或实施例。相反地,提供这些实施方式或实施例的目的是使对本发明的公开内容的理解更加透彻全面。
69.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式或实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”的可选范围包括两个或两个以上相关所列项目中任一个,也包括相关所列项目的任意的和所有的组合,所述任意的和所有的组合包括任意的两个相关所列项目、任意的更多个相关所列项目、或者全部相关所列项目的组合。
70.本发明中,“第一方面”、“第二方面”、“第三方面”等仅用于描述目的,不能理解为指示或暗示相对重要性或数量,也不能理解为隐含指明所指示的技术特征的重要性或数量。
71.本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
72.本发明中,涉及到数值区间,如无特别说明,则包括数值区间的两个端点。
73.本发明中涉及的百分比含量,如无特别说明,对于固液混合和固相
‑
固相混合均指质量百分比,对于液相
‑
液相混合指体积百分比。
74.本发明中涉及的百分比浓度,如无特别说明,均指终浓度。所述终浓度,指添加成分在添加该成分后的体系中的占比。
75.本发明中的温度参数,如无特别限定,既允许为恒温处理,也允许在一定温度区间内进行处理。所述的恒温处理允许温度在仪器控制的精度范围内进行波动。
76.需要说明的是,本发明的构建方法或者检测方法所涉步骤没有顺序限制。
77.本发明的下述实施例中所述试验方法,如无特别说明,均为常规方法;所述试剂和生物材料,如无特别说明,均可从商业途径获得。
78.第一方面,本发明提供一种乌蛇止痒丸的指纹图谱的构建方法,所述构建方法包括如下步骤:
79.取对照品和乌蛇止痒丸样品,分别制备对照品溶液和供试品溶液;采用高效液相色谱法对所述对照品溶液和所述供试品溶液进行检测,获得相应的检测图谱和对照图谱;将所述检测图谱导入中药色谱指纹图谱相似度评价系统进行分析,生成标准指纹图谱,参照所述对照图谱对所述标准指纹图谱的色谱峰进行指认,获得乌蛇止痒丸的指纹图谱;所述对照品包含人参皂苷rg1、人参皂苷re、人参皂苷rb1、人参皂苷rb2、人参皂苷rb3、人参皂苷rc、人参皂苷f1、人参皂苷rd、人参皂苷f2、人参皂苷rg2、人参皂苷rg3、5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、亥茅酚苷、蛇床子素、欧前胡素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸黄柏碱、盐酸药根碱、丹皮酚、芍药苷、丹皮酚原苷、苦参碱、氧化苦参碱、槐果碱、氧化槐果碱、木兰花碱、掌叶防已碱、柠檬苦素、黄柏酮、胆红素和二氢欧山芹醇当归酸酯中的至少一种;检测所采用条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲酸水溶液,流动相b为乙腈,采用梯度洗脱。
80.本发明提供的构建方法中,所述对照品包含人参皂苷rg1、人参皂苷re、人参皂苷rb1、人参皂苷rb2、人参皂苷rb3、人参皂苷rc、人参皂苷f1、人参皂苷rd、人参皂苷f2、人参皂苷rg2、人参皂苷rg3、5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、亥茅酚苷、蛇床子素、欧前胡素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸黄柏碱、盐酸药根碱、丹皮酚、芍药苷、丹皮酚原苷、苦参碱、氧化苦参碱、槐果碱、氧化槐果碱、木兰花碱、掌叶防已碱、柠檬苦素、黄柏酮、胆红素和二氢欧山芹醇当归酸酯中1种、2种、3种、4种、5种、6种、7种、8种、9种、10种、11种、12种、13种、14种、15种、16种、17种、18种、19种、20种、21种、22种、23种、24种、25种、26种、27种、28种、29种、30种、31种、32种、33种、34种、35种、36种或者37种。
81.在其中一个示例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比为5%;5min~35min,所述流动相b的体积百分比由5%升高至20%;35min~40min,所述流动相b的体积百分比为20%;40min~48min,所述流动相b的体积百分比由20%升高至23%;48min~60min,所述流动相b的体积百分比由23%升高至35%;60min~90min,所述流动相b的体积百分比由35%升高至55%;90min~105min,所述流动相b的体积百分比由55%升高至95%;105min~115min,所述流动相b的体积百分比为95%。
82.本发明提供的构建方法中,如所述流动相a选用水或甲酸水溶液,所述甲酸水溶液
中甲酸的体积百分比可以控制在0%~1.0%且不等于0%的范围,例如0.05%、0.1%、0.15%、0.2%、0.25%、0.3%、0.35%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%。考虑色谱柱对酸性流动相的耐受性及操作的可重复性,优选地,所述甲酸水溶液中甲酸的体积百分比可以控制在0%~0.15%且不等于0%的范围。
83.本发明提供的构建方法中,所述固定相中填料的粒径为1.5μm~3μm,例如可以为1.9μm、2.5μm、3μm,可选用的色谱柱包括但不限于thermofisher hypersil gold c18(150mm
×
2.1mm,1.9μm)、thermofisher hypersil gold c8(150mm
×
2.1mm,3μm)和waters xselect csh c18(150mm
×
3mm,2.5μm)。优选地,本发明选用c18色谱柱,例如hypersil gold c18色谱柱,其对乌蛇止痒丸中的特征峰的选择性更强,分离度和峰形更好。
84.在其中一个示例中,检测所采用条件还包括:波长为230nm~300nm。例如所述波长包括但不限于:230nm、235nm、240nm、245nm、250nm、254nm、260nm、265nm、270nm、275nm、280nm、285nm、290nm、295nm、300nm。优选地,所述波长为250nm~260nm,由此,获得谱图峰信息量较多。
85.在其中一个示例中,检测所采用条件还包括:流速为0.1ml/min~0.6ml/min,例如所述流速包括但不限于:0.1ml/min、0.2ml/min、0.3ml/min、0.35ml/min、0.4ml/min、0.45ml/min、0.5ml/min、0.6ml/min。
86.在其中一个示例中,检测所采用条件还包括:柱温为20℃~50℃,例如所述柱温包括但不限于:20℃、25℃、30℃、35℃、37℃、38℃、39℃、40℃、41℃、42℃、43℃、45℃、50℃。
87.在其中一个示例中,检测所采用条件还包括:进样量为1μl~6μl,例如所述进样量包括但不限于:1μl、2μl、3μl、4μl、5μl、6μl。
88.在其中一个示例中,制备所述对照品溶液采用的溶解溶剂含甲醇。
89.在其中一个示例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述乌蛇止痒丸样品进行提取,收集提取液。
90.在其中一个示例中,所述提取溶剂包含甲醇的体积百分比为10%~100%,例如可以为甲醇体积百分比为10~100%且不取100%(例如10%、20%、30%、40%、50%、60%、70%、80%、90%等)的甲醇水溶液,也可以为甲醇。
91.在其中一个示例中,提取的方式采用超声提取。优选地,超声提取的条件包括:功率为130w~300w(例如为130w、135w、140w、160w、180w、200w、210w、220w、230w、240w、250w、260w、270w、280w、290w、300w),频率为40khz~45khz(例如40khz、42khz、44khz),时长为10min~120min(例如10min、15min、20min、25min、30min、35min、40min、45min、50min、52min、55min、57min、58min、59min、60min、61min、62min、63min、65min、70min、75min、80min、90min、100min、110min、120min)。
92.在其中一个示例中,收集所述提取液的方式采用离心或者过滤。离心的条件包括10000rpm~15000rpm下离心5min~15min,例如10000rpm下15min、15000rpm下5min、14000rpm下10min。过滤可以采用例如0.45μm或者0.22μm的滤膜。
93.在其中一个示例中,所述特征图谱包含34个特征峰,其中,7号峰为升麻素苷,9号峰为升麻素、14号峰为5
‑
o
‑
甲基维斯阿米醇苷,15号峰为药根碱,20号峰为小檗碱,21号峰为巴马汀,22号峰为丹皮酚,23号峰为佛手柑内酯,25号峰为花椒毒素,30号峰为花椒毒酚,32号峰为蛇床子素。
94.本发明提供一种乌蛇止痒丸的检测方法,该检测方法利用液相色谱法,并通过采用合适的流动相进行梯度洗脱,能够全面地提供乌蛇止痒丸整体化学成分信息,使乌蛇止痒丸的质量控制更为准确和全面。
95.第二方面,本发明提供一种乌蛇止痒丸的检测方法,所述检测方法包括如下步骤:
96.取待测样品,制备供试品溶液,采用高效液相色谱法对所述供试品溶液进行检测,并将所得检测图谱与如上构建获得的指纹图谱进行比较;
97.检测所采用条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲酸水溶液,流动相b为乙腈,采用梯度洗脱。
98.在其中一个示例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比为5%;5min~35min,所述流动相b的体积百分比由5%升高至20%;35min~40min,所述流动相b的体积百分比为20%;40min~48min,所述流动相b的体积百分比由20%升高至23%;48min~60min,所述流动相b的体积百分比由23%升高至35%;60min~90min,所述流动相b的体积百分比由35%升高至55%;90min~105min,所述流动相b的体积百分比由55%升高至95%;105min~115min,所述保持流动相b的体积百分比为95%。
99.在其中一个示例中,所述甲酸水溶液含甲酸的体积百分比为0.1%~1.0%。
100.在其中一个示例中,所述固定相中填料的粒径为1.5μm~3μm。
101.在其中一个示例中,检测所采用条件还包括:波长为230nm~300nm;或/和,流速为0.1ml/min~0.6ml/min;或/和,柱温为20℃~50℃;或/和,进样量1μl~6μl。
102.在其中一个示例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述待测样品进行提取,收集提取液。
103.在其中一个示例中,所述提取溶剂中甲醇的体积百分比为10%~100%。
104.在其中一个示例中,提取的方式采用超声提取。
105.在其中一个示例中,收集所述提取液的方式采用离心或者过滤。
106.可以理解地,上述乌蛇止痒丸的检测方法中的供试品溶液的制备以及检测所述供试品溶液的条件与乌蛇止痒丸指纹图谱构建方法中相同。在此不再赘述。
107.仅通过定性指纹分析对乌蛇止痒丸的综合质量评价是不够的。因此,本发明还基于uplc
‑
ms/ms技术建立了乌蛇止痒丸中多成分的定量方法,并在本发明中选择了16种化学标志物进行定量。值得注意的是,这些化学指标被筛选并选择为感兴趣的分析物,因为它们被记录在中国药典(2020年版)或在文献中广泛报道。实际上,许多目标化合物的含量都在dad检测范围内。然而,为了获得良好的基线分离,同时准确定量复杂乌蛇止痒丸样品中的16种化合物,一旦采用dad检测,复杂而乏味的优化过程和过长的分析时间是不可避免的。相反,串联ms/ms方法的优化效率更高,只需要对离子对、锥电压(cv)和碰撞能量(ce)进行优化,而不是复杂且耗时的色谱基线分离。
108.第三方面,本发明提供一种乌蛇止痒丸的多成分含量检测方法,所述检测方法包括如下步骤:
109.取对照品和待测样品,分别制备对照品溶液和供试品溶液;采用高效液相色谱质谱联用法对所述对照品溶液和所述供试品溶液进行检测,并根据检测所述对照品溶液所得图谱和检测所述供试品溶液所得图谱中各特征峰的峰面积,确定所述乌蛇止痒丸中对应成分的含量;
110.所述对照品包含苦参碱、槐果碱、氧化苦参碱、升麻素、盐酸黄柏碱、木兰花碱、升麻素苷、盐酸小檗碱、盐酸巴马汀、丹皮酚、花椒毒素、佛手柑内酯、人参皂苷rb1、人参皂苷rb2、人参皂苷rc和二氢欧山芹醇当归酸酯中的至少一种;
111.所述高效液相色谱质谱联用法中,液相色谱条件包括:固定相采用c8色谱柱或者c18色谱柱,流动相a为水或者甲酸水溶液,流动相b为乙腈,采用梯度洗脱。
112.本发明提供的多成分含量检测方法,所述对照品包含苦参碱、槐果碱、氧化苦参碱、升麻素、盐酸黄柏碱、木兰花碱、升麻素苷、盐酸小檗碱、盐酸巴马汀、丹皮酚、花椒毒素、佛手柑内酯、人参皂苷rb1、人参皂苷rb2、人参皂苷rc和二氢欧山芹醇当归酸酯中的1种、2种、3种、4种、5种、6种、7种、8种、9种、10种、11种、12种、13种、14种、15种或者16种。
113.在其中一个示例中,梯度洗脱的程序包括:0min~5min,所述流动相b的体积百分比由5%升高至25%;5min~7min,所述流动相b的体积百分比由25%升高至37%;7min~8min,所述流动相b的体积百分比为37%;8min~10min,所述流动相b的体积百分比由37%升高至55%;10min~12min,所述流动相b的体积百分比由55%升高至95%;12min~14min,所述流动相b的体积百分比为95%;14min~15min,所述流动相b的体积百分比由95%下降至5%;15min~17min,所述流动相b的体积百分比为5%。
114.在其中一个示例中,所述固定相中填料的粒径为1.5μm~3μm。例如可以为1.9μm、2.5μm、3μm,可选用的色谱柱包括但不限于hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher)、hypersil gold c8(150mm
×
2.1mm,3μm,thermo fisher)和acquitytm uplc beh c18(100mm
×
2.1mm,1.7μm,沃特世)。优选地,本发明选用c18色谱柱,例如hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher),在优选色谱柱上,两种相似的化合物g
‑
rb2和g
‑
rc可以在hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher)色谱柱上实现基线分离,其他14种目标分析物也显示出在同一色谱柱上的最佳保留行为和最高强度。
115.在其中一个示例中,所述液相色谱条件还包括:流速为0.1ml/min~0.6ml/min,例如所述流速包括但不限于:0.1ml/min、0.2ml/min、0.3ml/min、0.35ml/min、0.4ml/min、0.45ml/min、0.5ml/min、0.6ml/min。
116.在其中一个示例中,所述液相色谱条件还包括:柱温为20℃~50℃,例如所述柱温包括但不限于:20℃、25℃、30℃、35℃、37℃、38℃、39℃、40℃、41℃、42℃、43℃、45℃、50℃。
117.在其中一个示例中,检测所采用条件还包括:进样量为1μl~6μl,例如所述进样量包括但不限于:1μl、2μl、3μl、4μl、5μl、6μl。
118.在其中一个示例中,所述高效液相色谱质谱联用法中,质谱条件包括:esi正离子模式,毛细管电压:2.5kv~3.5kv,源温度:145℃~155℃,脱溶剂温度:335℃~345℃,脱溶剂气流:640l/h~660l/h,锥形气流:25l/h~35l/h,碰撞气体:氩,脱溶剂气体:氮气。
119.每个化合物的mrm跃迁、锥孔电压(cv)、碰撞能量(ce)等质谱参数见表5在其中一个示例中,制备所述对照品溶液采用的溶解溶剂含有甲醇。
120.在其中一个示例中,制备所述供试品溶液的步骤包括:用提取溶剂对所述待测样品进行提取,收集提取液。
121.在其中一个示例中,所述提取溶剂含甲醇的体积百分比为10%~100%。所述提取
溶剂包含甲醇的体积百分比为10%~100%,例如可以为甲醇体积百分比为10~100%且不取100%(例如10%、20%、30%、40%、50%、60%、70%、80%、90%等)的甲醇水溶液,也可以为甲醇。
122.在其中一个示例中,提取的方式采用超声提取。优选地,超声提取的条件包括:功率为130w~300w(例如为130w、135w、140w、160w、180w、200w、210w、220w、230w、240w、250w、260w、270w、280w、290w、300w),频率为40khz~45khz(例如40khz、42khz、44khz、45khz),时长为10min~120min(例如10min、15min、20min、25min、30min、35min、40min、45min、50min、52min、55min、57min、58min、59min、60min、61min、62min、63min、65min、70min、75min、80min、90min、100min、110min、120min)。
123.在其中一个示例中,收集所述提取液的方式采用离心或者过滤。离心的条件包括10000rpm~15000rpm下离心5min~15min,例如10000rpm下15min、15000rpm下5min、14000rpm下10min。过滤可以采用例如0.45μm或者0.22μm的滤膜。
124.本发明提供的上述乌蛇止痒丸的多成分含量检测方法,可在13min内同时测定乌蛇止痒丸中的16种化学标志物,是一种快速的乌蛇止痒丸的质量控制方法。
125.如下为具体的实施例,如无特别说明,实施例中采用的原料均为市售获得。
126.实施例1、乌蛇止痒丸指纹图谱构建
127.本实施例提供一种乌蛇止痒丸的指纹图谱的构建方法,该构建方法包括以下步骤:
128.1.实验仪器与试药
129.仪器:ultimate 3000快速分离液相色谱系统(美国戴安公司),包括:hpg
‑
3400rs二元高压梯度泵,wps
‑
3000trs自动进样器,tcc
‑
3000rs柱温箱,rs variable可变波长紫外检测器,corona veo rs电雾式检测器(cad)以及变色龙色谱管理软件(sr7.2);milli
‑
q超纯水处理系统(美国millipore公司);高速低温离心机(美国thermofisher公司);ltq
‑
orbitrap xl组合式高分辨质谱。
130.试剂:乙腈、甲醇、甲酸均为色谱纯试剂,磷酸为分析纯试剂,实验用超纯水(18.2mohm.cm)由milli
‑
q超纯水系统制得。
131.对照品:人参皂苷rg1、人参皂苷re、人参皂苷rb1、人参皂苷rb2、人参皂苷rb3、人参皂苷rc、人参皂苷f1、人参皂苷rd、人参皂苷f2、人参皂苷rg2、人参皂苷rg3、5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、亥茅酚苷、蛇床子素、欧前胡素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸黄柏碱、盐酸药根碱、丹皮酚、芍药苷、丹皮酚原苷、苦参碱、氧化苦参碱、槐果碱、氧化槐果碱、木兰花碱、掌叶防已碱、柠檬苦素、黄柏酮、胆红素、二氢欧山芹醇当归酸酯均购自于成都曼斯特生物科技有限公司且纯度都≥95%,所有对照品在使用前均通过紫外吸收光谱、高分辨质谱以及多级碎片数据进行确证。
132.实验样品:试验中所有批次乌蛇止痒丸成品样品均由广州白云山中一药业有限公司提供,详见表1。
133.表1、乌蛇止痒丸样品
134.编号样品批次编号样品批次s1z04003s9z04007s2y04004ms10z04001
s3z04011s11z04014s4y04014s12z04016s5y04001ms13z04009s6y04011s14x00029s7z04005s15yn0001s8y04007ms16y00003
135.2.方法
136.2.1色谱条件
137.色谱柱为thermofisher hypersil gold c18(150mm
×
2.1mm,1.9μm)。
138.流动相a为0.1%甲酸水,b为乙腈,梯度洗脱:0min~5min,5%b;5min~35min,5%~20%b;35min~40min,20%b;40min~48min,20%~23%b;48min~60min,23%~35%b;60min~90min,35%~55%b;90min~105min,55%~95%b,105min~115min,95%b。
139.流速:0.3ml/min;柱温:40℃;进样量:4μl。
140.二极管阵列检测器(dad)检测条件,检测波长:280nm、254nm;采样频率:5hz。
141.cad检测器检测条件,雾化室温度:35℃;采样频率:10hz。
142.2.2供试品溶液制备
143.精密称取乌蛇止痒丸粉末约1g,精密称定,置具塞锥形瓶中,精密加入70%甲醇20ml,密塞,称定质量,超声处理(branson 5510;功率/频率,135w/42khz)30min,放冷,称重,用70%甲醇补足失重,摇匀,静置,取上清液于14000rpm高速离心10min,取上清液置于液相色谱进样瓶,即得。
144.2.3对照品溶液制备
145.分别精密称取人参皂苷rg1、人参皂苷re、人参皂苷rb1、人参皂苷rb2、人参皂苷rb3、人参皂苷rc、人参皂苷f1、人参皂苷rd、人参皂苷f2、人参皂苷rg2、人参皂苷rg3、5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、亥茅酚苷、蛇床子素、欧前胡素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸黄柏碱、盐酸药根碱、丹皮酚、芍药苷、丹皮酚原苷、苦参碱、氧化苦参碱、槐果碱、氧化槐果碱、木兰花碱、掌叶防已碱、柠檬苦素、黄柏酮、胆红素、二氢欧山芹醇当归酸酯对照品适量,加色谱甲醇溶解并定容至2ml作为标准储备液,并置于4℃的冰箱中保存备用。
146.2.4数据处理
147.实验中图谱均由变色龙色谱管理软件(sr7.2)处理并导出;所得导出后样品图谱以“中药色谱指纹图谱相似度评价系统2004a版”(国家药典委员会)进行指纹图谱分析,计算样品相似度。
148.2.5标准指纹图谱的建立
149.2.5.1指纹图谱的检测
150.取16个批次的乌蛇止痒丸样品,按如上2.1项下所述的色谱条件进行分析,结果见图1。
151.2.5.2对照图谱和共有峰的标定
152.将16批次乌蛇止痒丸样品的hplc指纹图谱导入《中药色谱指纹图谱相识度评价系统2004a》进行数据处理,建立共有模式指纹图谱,即乌蛇止痒丸标准指纹图谱,见图2。
153.根据16批乌蛇止痒丸样品的测定结果确定34个共有峰。选择25号峰(花椒毒素)为参照峰,将其保留时间和峰面积均设为1.0,计算各共有峰的相对保留时间及相对峰面积分别列于表2和表3。
154.表2、16批次乌蛇止痒丸指纹图谱共有峰的相对保留时间
[0155][0156][0157]
表2(续)、16批次乌蛇止痒丸指纹图谱共有峰的相对保留时间
[0158]
[0159][0160]
表3、16批次乌蛇止痒丸指纹图谱共有峰的相对保留峰面积
[0161]
[0162][0163]
表3(续)、16批次乌蛇止痒丸指纹图谱共有峰的相对保留峰面积
[0164]
[0165][0166]
2.5.3相似度评价
[0167]
将16批次乌蛇止痒丸样品的hplc指纹图谱导入《中药色谱指纹图谱相识度评价系统2004a版》,生成乌蛇止痒丸标准指纹图谱,在分析检验模式下,计算16批次乌蛇止痒丸样品标准指纹图谱的相似度,结果见表4,所有代表性样品与标准指纹图谱的相似度均在0.930以上。
[0168]
表4、指纹图谱相似性
[0169][0170]
表4(续)、指纹图谱相似性
[0171]
[0172][0173]
2.5.4色谱峰指认及归属分析
[0174]
经与检测对照品溶液所得对照图谱比对和质谱确认后,表明标准指纹图谱特征峰中7号峰为升麻素苷(防风),9号峰为升麻素(防风)、14号峰为5
‑
o
‑
甲基维斯阿米醇苷(防风),15号峰为药根碱(关黄柏),20号峰为小檗碱(关黄柏),21号峰为巴马汀(关黄柏),22号峰为丹皮酚(牡丹皮),23号峰为佛手柑内酯(防风、蛇床子),25号峰为花椒毒素(防风、蛇床子),30号峰为花椒毒酚(蛇床子),32号峰为蛇床子素(蛇床子、当归)。
[0175]
2.6方法学考察
[0176]
2.6.1精密度
[0177]
按2.2项下制备方法制备供试品溶液,连续进样6次,进行uplc分析,结果表明,各共有峰相对峰面积和相对保留时间的rsd值分别小于4.95%和0.72%,表明仪器精密度良好,符合指纹图谱要求。
[0178]
2.6.2稳定性
[0179]
按2.2项下制备方法制备供试品溶液,分别在0h、3h、6h、9h、12h和24h时进行uplc分析,结果表明,各共有峰相对峰面积和相对保留时间的rsd值分别小于4.96%和0.93%,表明供试品溶液在24h内稳定性良好。
[0180]
2.6.3重复性
[0181]
取同一批次样品6份,按2.2项下制备方法制备供试品溶液,进行uplc分析,结果表明,各共有峰相对峰面积和相对保留时间的rsd值分别小于4.90%和0.90%,表明该方法重复性较好。
[0182]
实施例2、乌蛇止痒丸的指纹图谱构建方法的条件考察
[0183]
本实施例涉及乌蛇止痒丸的指纹图谱构建方法的条件考察,包括如下:
[0184]
1.供试品溶液制备工艺考察
[0185]
1.1提取溶剂考察
[0186]
相对于实施例1,保持其它条件不变,比较了10%、40%、70%和100%甲醇共4种提取溶剂体系的超声提取效率。
[0187]
结果如图3所示,随着提取溶剂中甲醇浓度的升高,检测得到的色谱峰也逐步增多,尤其是极性较弱组分峰,同时色谱峰强度也逐步升高,表明提取效率的提高,并以70%甲醇为提取溶剂时,色谱峰数量最多,继续升高甲醇浓度,色谱峰强度有所降低。因此,选用70%甲醇作为乌蛇止痒丸指纹图谱样品的提取溶剂。本发明采用70%甲醇超声提取30分钟
的优化方案用于指纹图谱构建、乌蛇止痒丸的定性检测以及乌蛇止痒丸中多成分的定量研究(见实施例3)。
[0188]
1.2提取时间考察
[0189]
相对于实施例1,保持其它条件不变,比较超声提取时间分别为10min、30min、60min、90min条件下的提取效率。
[0190]
结果如图4所示,随着提取时间的增加,各色谱峰响应强度也逐步增强,当提取时间为30min时,色谱峰强度达到最高,并且提取时间为60min和90min时,色谱峰强度无明显提高。因此。以提取效率高同时节约时间为原则,优选30min作为乌蛇止痒丸指纹图谱样品的超声提取时间。
[0191]
2.色谱条件考察
[0192]
利用uplc对供试品溶液进行分离检测,优化分离条件,包括检测器类型、色谱柱种类、色谱柱温度、流动相组成、流动相添加剂等,使被分析样品能在最短的时间内尽可能多的得到基线分离的色谱峰。
[0193]
2.1色谱柱考察
[0194]
相对于实施例1,保持其它条件不变,比较三根不同类型的色谱柱的分离情况,包括thermofisher hypersil gold c18(150mm
×
2.1mm,1.9μm)、thermofisher hypersil gold c8(150mm
×
2.1mm,3μm)和waters xselect csh c18(150mm
×
3mm,2.5μm)。
[0195]
结果如图5所示,采用hypersil gold c18色谱柱时选择性更强,分离度和峰形更好,因此选择hypersil gold c18色谱柱进行后续指纹图谱研究。
[0196]
2.2流动相考察
[0197]
相对于实施例1,保持其它条件不变,比较不同流动相的组成的分离情况,包括甲醇
‑
水、乙腈
‑
水、乙腈
‑
0.1%甲酸水(体积浓度)、乙腈
‑
0.2%甲酸水(体积浓度)、乙腈
‑
0.4%甲酸水(体积浓度)。
[0198]
结果如图6和图7所示,结果表明采用乙腈
‑
水作为流动相进行梯度程序洗脱时基线平稳,色谱峰更多,峰型更好,并且灵敏度更高。因此,选择乙腈
‑
水体系进行研究。添加一定浓度的甲酸添加剂后,5min~15min内色谱峰信号响应有一定增强,在不同添加剂浓度条件下,色谱图无明显差异。同时,考虑色谱柱对酸性流动相的耐受性及实验的可重复性,因此采用乙腈
‑
0.1%甲酸水作为流动相进行梯度洗脱分析。
[0199]
2.3柱温考察
[0200]
相对于实施例1,保持其它条件不变,比较不同色谱柱柱温对样品分离的影响,包括35℃、40℃、45℃。
[0201]
结果见图8,结果显示,色谱柱温度为35℃、40℃、45℃时,各色谱峰分离效果类似,且均能得到较好的峰形。鉴于色谱柱寿命及实验的可重复性,本实验选择色谱柱温度为40℃。
[0202]
2.4检测器的选择
[0203]
由于乌蛇止痒丸由多达11味中药组成,其化学成分数量、性质和种类很多,因此本发明建立的指纹图谱要尽可能多的得到化学成分的信息。为了选择更加灵敏和通用型的检测器,本实验考察了dad紫外检测器(设置多波长检测)和cad检测器进行了比较。
[0204]
结果如图9所示,在cad检测器下,色谱图基线较高且不平,各色谱峰存在漂移情
况。本发明尝试利用dad检测器进行样品分析,结果显示在254nm紫外检测下,图谱中强极性成分信号响应强度极高,而中等极性和弱极性成分峰数目稍少,但信号强度高,基线平稳,分离度良好;同时改变检测波长为280nm后,紫外吸收较差,色谱图中峰信号强度明显降低。相比于cad检测器,dad紫外检测器结果灵敏度更高,且在254nm紫外检测下,峰强度最高。因此,本发明选择dad紫外检测器进行后续乌蛇止痒丸指纹图谱研究。
[0205]
2.5梯度程序的优化
[0206]
乌蛇止痒丸由11味中药组成,化学成分数量庞大和种类繁多,同时指纹图谱要求尽可能将样品中所含化学成分分离表征,因此本发明基于uplc
‑
dad技术,使用1.9μm粒径超高效液相色谱柱,对样品进行色谱分析。同时,由于所含化学成分的性质不一,本发明优化梯度洗脱程序以求在最短的时间内分离得到最大数量的色谱峰(参见实施例1第2.1项下梯度洗脱程序),最后在115min内完成了样品的分析(图10)。
[0207]
除了最终采纳的最优程序,本实施例还使用了其它梯度洗脱程序进行分析,例如:流动相a为水,b为甲醇,梯度洗脱:0min~15min,5%~20%b;15min~45min,20%~30%b;45min~120min,30%~75%b,结果显示,在此梯度洗脱程序下,色谱峰不能得到很好的分离(图11)。
[0208]
实施例3、用乌蛇止痒丸指纹图谱对待测样品进行检测
[0209]
本实施例提供一种乌蛇止痒丸的检测方法,该检测方法包括以下步骤:
[0210]
1.实验仪器与试药
[0211]
仪器:ultimate 3000快速分离液相色谱系统(美国戴安公司),包括:hpg
‑
3400rs二元高压梯度泵,wps
‑
3000trs自动进样器,tcc
‑
3000rs柱温箱,rs variable可变波长紫外检测器,以及变色龙色谱管理软件(sr7.2);milli
‑
q超纯水处理系统(美国millipore公司);高速低温离心机(美国thermofisher公司);
[0212]
试剂:乙腈、甲醇、甲酸均为色谱纯试剂,实验用超纯水(18.2mohm.cm)由milli
‑
q超纯水系统制得。
[0213]
对照品:5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、蛇床子素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸药根碱、丹皮酚均购自于成都曼斯特生物科技有限公司且纯度都≥95%,所有对照品在使用前均通过紫外吸收光谱、高分辨质谱以及多级碎片数据进行确证。
[0214]
实验样品:y04006m批次乌蛇止痒丸样品由广州白云山中一药业有限公司提供。
[0215]
2.方法
[0216]
2.1色谱条件
[0217]
色谱柱为thermofisher hypersil gold c18(150mm
×
2.1mm,1.9μm)。
[0218]
流动相a为0.1%甲酸水,b为乙腈,梯度洗脱:0min~5min,5%b;5min~35min,5%~20%b;35min~40min,20%b;40min~48min,20%~23%b;48min~60min,23%~35%b;60min~90min,35%~55%b;90min~105min,55%~95%b,105min~115min,95%b。
[0219]
流速:0.3ml/min;柱温:40℃;进样量:4μl。
[0220]
二极管阵列检测器(dad)检测条件,检测波长:254nm;采样频率:5hz。
[0221]
2.2供试品溶液制备
[0222]
精密称取乌蛇止痒丸粉末约1g,精密称定,置具塞锥形瓶中,精密加入70%甲醇
20ml,密塞,称定质量,超声处理(branson 5510;功率/频率,135w/42khz)30min,放冷,称重,用70%甲醇补足失重,摇匀,静置,取上清液于14000rpm高速离心10min,取上清液置于液相色谱进样瓶,即得。
[0223]
2.3对照品溶液制备
[0224]
分别精密称取5
‑
o
‑
甲基维斯阿米醇苷、升麻素苷、升麻素、蛇床子素、花椒毒酚、花椒毒素、佛手柑内酯、盐酸小檗碱、盐酸巴马汀、盐酸药根碱、丹皮酚对照品适量,加色谱甲醇溶解并定容至2ml作为标准储备液,并置于4℃的冰箱中保存备用。
[0225]
2.4数据处理
[0226]
实验中图谱均由变色龙色谱管理软件(sr7.2)处理并导出;所得导出后样品图谱以“中药色谱指纹图谱相似度评价系统2004a版”(国家药典委员会)进行指纹图谱分析,计算样品相似度。
[0227]
2.5指纹图谱的检测
[0228]
取所述供试品溶液和所述对照品溶液,按如上所述的2.1项下色谱条件进行分析,结果见图12。
[0229]
2.6相似度评价
[0230]
将y04006m批次乌蛇止痒丸样品的检测图谱导入《中药色谱指纹图谱相识度评价系统2004a版》,计算检测图谱与实施例1构建的具有标示性的乌蛇止痒丸的指纹图谱相的相似度,结果显示,该检测图谱与指纹图谱的相似度为0.985。
[0231]
2.7色谱峰指认
[0232]
经与检测对照品溶液所得的对照图谱比对,表明指纹图谱特征峰中7号峰为升麻素苷(防风),9号峰为升麻素(防风)、14号峰为佛手柑内酯(防风),15号峰为盐酸药根碱(关黄柏),20号峰为盐酸小檗碱(关黄柏),21号峰为巴马汀(关黄柏),22号峰为丹皮酚(牡丹皮),23号峰为佛手柑内酯(防风、蛇床子),25号峰为花椒毒素(防风、蛇床子),30号峰为花椒毒酚(蛇床子),32号峰为蛇床子素(蛇床子、当归)。所得结果与前述指纹图谱一致,说明此指纹图谱及构建该指纹图谱的色谱条件可用于乌蛇止痒丸的检测。
[0233]
实施例4、乌蛇止痒丸的多成分含量检测方法
[0234]
1.实验仪器与试药
[0235]
仪器:ultimate 3000快速分离液相色谱系统(美国戴安公司),包括:hpg
‑
3400rs二元高压梯度泵,wps
‑
3000trs自动进样器,tcc
‑
3000rs柱温箱,rs variable可变波长紫外检测器,corona veo rs电雾式检测器(cad)以及变色龙色谱管理软件(sr7.2);milli
‑
q超纯水处理系统(美国millipore公司);高速低温离心机(美国thermofisher公司);ltq
‑
orbitrap xl组合式高分辨质谱。
[0236]
试剂:乙腈、甲醇、甲酸均为色谱纯试剂,磷酸为分析纯试剂,实验用超纯水(18.2mohm.cm)由milli
‑
q超纯水系统制得。超纯水(18.2m.cm at 25℃)由milli
‑
q纯化系统(millipore,bedford,ma,usa)制备。lc
‑
ms级乙腈和甲醇购自fisher scientific(英国拉夫堡)。其他化学品均为分析级。
[0237]
对照品:苦参碱、槐果碱、氧化苦参碱、升麻素、盐酸黄柏碱、木兰花碱、升麻素苷、盐酸小檗碱、盐酸巴马汀、丹皮酚、花椒毒素、佛手柑内酯、人参皂苷rb1、人参皂苷rb2、人参皂苷rc、二氢欧山芹醇当归酸酯由成都曼斯特生物科技有限公司(成都,四川,中国)提供,
纯度高于98%。
[0238]
实验样品:试验中乌蛇止痒丸成品样品由广州白云山中一药业有限公司提供。详见表1。
[0239]
2.方法和结果
[0240]
2.1检测条件
[0241]
采用了连接到xevo tqd三重四极杆质谱仪(沃特世公司,曼彻斯特,英国)的acquity i
‑
class uplc系统。
[0242]
hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher)色谱柱用于分离保持在40℃的目标分析物。流速为0.3ml/min,流动相由含0.1%甲酸(a)和乙腈(b)的水组成,梯度洗脱程序如下:0min~5min,5%~25%b;5min~7min,25%~37%b;7min~8min,37%b;8min~10min,37%~55%b;10min~12min,55%~95%b;12min~14min,95%b;14min~15min,95%~5%b;15min~17min,5%b。注射体积为2μl。
[0243]
检测在正esi离子模式下进行,最佳ms参数设置如下:毛细管电压,3.0kv;源温度,150℃;脱溶剂温度,350℃;脱溶剂气流量,650l/h;锥形气流,30l/h。n2和高纯氩(ar)分别作为去溶剂化气体和碰撞气体。mrm模式用于多成分定量,详细的ms/ms检测参数汇总于表5。
[0244]
表5、乌蛇止痒丸中16种分析物的保留时间(rt)、mrm跃迁、锥孔电压(cv)、碰撞能量(ce)
[0245][0246][0247]
masslynx 4.2软件(waters corp.,milford,ma,usa)用于仪器控制和数据分析。
[0248]
2.2供试品溶液的制备
[0249]
精确称取1g乌蛇止痒丸的细粉样品,在20ml 70%(v/v)甲醇水溶液中通过超声水浴(branson 8510,250w,44khz)在室温下萃取30min。然后,将上清液以14000rpm离心10min,收集上清液进行分析。
[0250]
2.3对照品溶液制备
[0251]
分别精密称取苦参碱、槐果碱、氧化苦参碱、升麻素、盐酸黄柏碱、木兰花碱、升麻素苷、盐酸小檗碱、盐酸巴马汀、丹皮酚、花椒毒素、佛手柑内酯、人参皂苷rb1、人参皂苷
rb2、人参皂苷rc、二氢欧山芹醇当归酸酯,加色谱甲醇溶解并定容至2ml作为标准储备液,并置于4℃的冰箱中保存备用。在分析前用甲醇稀释至合适的浓度。
[0252]
2.3结果
[0253]
分别将16批次的乌蛇止痒丸样品制备的供试品溶液和对照品溶液上样至仪器,在第2.1项所示检测条件下检测,测定不同批次乌蛇止痒丸样品中16种目标化合物的含量,一式三份,统计结果列于表6中。本发明提供的乌蛇止痒丸多成分含量检测方法,用于16种化合物的总含量范围为3323.3μg/g~4445.0μg/g,其中苦参碱的含量(1126.8μg/g~1612.5μg/g)远大于其他15种化合物。相比之下,g
‑
rb1(14.9μg/g~22.7μg/g)、g
‑
rb2(19.6μg/g~31.7μg/g)和g
‑
rc(16.7μg/g~30.0μg/g)的含量相对较低。同时,二氢欧山芹醇当归酸酯被认为是含量最低的化合物。如图13所示,乌蛇止痒丸样品中16种目标化合物通过建立的uplc
‑
ms/ms方法实现了良好的分离,无其他干扰,可作为uplc
‑
dad指纹图谱的适当补充,用于乌蛇止痒丸的综合质量评价。
[0254]
表6、不同批次乌蛇止痒丸样品中16种目标分析物的含量(μg/g,平均值
±
sd)
[0255][0256][0257]
表6(续)、不同批次乌蛇止痒丸样品中16种目标分析物的含量(μg/g,平均值
±
sd)
[0258][0259]
3含量检测过程中的uplc
‑
ms/ms条件的优化
[0260]
针对uplc
‑
ms/ms分析优化了固定相、流动相、梯度洗脱程序和相关的ms/ms参数。
[0261]
色谱柱的选择是对目标分析物(尤其是g
‑
rb2和g
‑
rc)实现良好保留和高分辨率的关键因素,它们在rp色谱柱上表现出相似的保留行为,并且具有相同的ms2碎片离子。因此,测试了具有不同固定相的rp色谱柱,包括hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher)、hypersil gold c8(150mm
×
2.1mm,3μm,thermo fisher)和acquitytm uplc beh c18(100mm
×
2.1mm,1.7μm,沃特世)。因此,两种相似的化合物g
‑
rb2和g
‑
rc可以在hypersil gold c18(150mm
×
2.1mm,1.9μm,thermo fisher)色谱柱上实现基线分离,其他14种目标分析物也显示出在同一色谱柱上的最佳保留行为和最高强度(图14)。
[0262]
基于优化的指纹图谱构建条件(实施例2),乙腈(流动相b)
‑
0.1%甲酸(流动相a,0.1%甲酸水溶液)也用于uplc
‑
ms/ms分析作为最佳流动相。
[0263]
鉴于乌蛇止痒丸中目标分析物的广泛极性范围,采用了优化的梯度洗脱程序,如第2.1项下的梯度洗脱程序。
[0264]
为了获得令人满意的灵敏度和选择性,采用mrm模式进行定量分析。通过intelli start程序(waters corp.,milford,ma,usa)根据最高峰值信号和最低串扰或其他干扰的规则优化相关ms/ms参数,结果列于表5。uplc
‑
ms/ms条件下,所有16种目标化合物在mrm模
式下仅在13分钟内独立分离,无干扰,16种分析物在正离子模式下的典型ms/ms谱如图15所示。
[0265]
4uplc
‑
ms/ms分析的方法验证
[0266]
4.1线性、检测和定量下限
[0267]
分析一系列具有所需浓度的工作标准溶液以建立校准曲线。信噪比(s/n)为3和10时每种分析物的浓度分别定义为检测下限(llod)和定量(lloq)。
[0268]
通过将峰面积(y)与每种分析物的相应浓度(x)作图,获得16种目标化合物的校准曲线,显示出良好的线性,相关系数(r)高于0.995(表7)。lloq和llod分别在0.9ng/ml~23.8ng/ml和0.2ng/ml~10.3ng/ml的范围内。上述结果表明所构建的uplc
‑
ms/ms方法具有良好的可靠性和高灵敏度。
[0269]
4.2精密度、可重复性、稳定性和准确性
[0270]
相同的混合标准溶液测试6次进行精密度评估。用六个独立制备的样品评估了重复性。通过在一天内的6个时间点(0h、3h、6h、9h、12h和24h)分析相同的样品溶液来评估稳定性。计算相应峰面积的rsd值以评估上述测试。
[0271]
进行回收率实验以评估准确度。将已知量的单个化学对照品加入wzp样品中。按照建议的程序处理了六个强化样品。回收率(%)的计算公式如下:回收率(%)=(检出量
‑
原始量)/加标量
×
100%。
[0272]
就精密度而言,分析物的rsd在1.4%~7.1%的可接受范围内。重复性的rsd小于5.9%。16种目标化合物在24h内均稳定,其rsd范围为1.0%至5.6%。此外,所有16种成分的回收率均符合限度规定。
[0273]
表7、乌蛇止痒丸中16种目标分析物的校准曲线、线性范围、llod、lloq、回收率、精密度、重复性和稳定性
[0274]
[0275][0276]
随着中药方剂的广泛临床应用和进一步国际化,对中药方剂的质量控制和评价的要求也逐渐提高。本发明以uplc
‑
dad指纹图谱与多化合物定量分析相结合的综合策略为案例,优化并用于中药方剂乌蛇止痒丸的质量控制和评估。总共筛选了34个特征共同峰,用于评估来自16个批次的乌蛇止痒丸的化学特征的相似性。对于定量分析,采用优化的uplc
‑
ms/ms方法同时测定16种目标化合物的含量,具有可接受的灵敏度、精密度、重现性和准确度。对16批乌蛇止痒丸样品的数据分析表明,16个目标的总含量未观察到显着变化,表明乌蛇止痒丸样品的质量一致性良好。综上所述,这是首次将整体化学特征与显着目标化合物相结合,系统地评估乌蛇止痒丸的质量,为监测中药方剂的整体质量一致性提供了一种可行且有效的策略。
[0277]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。
[0278]
应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所述附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。