1.本发明涉及生物信息学领域,尤其涉及一种基于深度编码与多模态融合的预测新生抗原免疫原性的方法和系统。
背景技术:
2.免疫治疗已成为一种很有希望的癌症治疗策略。各种形式的免疫治疗可以增强免疫系统以抵抗癌症,或者使免疫系统更容易识别并摧毁癌细胞,或减慢其生长。有效的靶向免疫治疗需要精确的预测哪些癌症特异性新肽段最有可能引起免疫反应。
3.cd8 t细胞免疫反应是识别和杀死感染细胞和恶性肿瘤细胞的关键。过去的十年中癌症免疫治疗表明,利用增强cd8 t细胞的介导对癌细胞的控制和清除具有临床意义。在分子水平层面,cd8 t细胞对肽表位的识别基于一系列特定事件。首先,肽段被蛋白酶从源蛋白上切割,转运到内质网中并与hla
‑
i分子结合。稳定结合后,肽
‑
hla
‑
i(phla)复合物被呈现在细胞表面。随后,t细胞受体(tcr)可以与phla复合物结合,从而启动免疫突触的形成,并最终导致被感染或恶性细胞的死亡。
4.基于hla呈递
‑
cd8 t细胞识别原理的癌症疫苗是当今医学与药物学的热点问题。肿瘤疫苗教导免疫系统将传染性病原体或癌细胞识别为需要消除的外来物质。癌细胞表面存在特殊的蛋白质,通过靶向这些蛋白质,免疫系统可以特异性地消除癌细胞,同时不伤害正常的细胞。此外,疫苗还能防止癌症复发,清除治疗后残留的癌细胞。肿瘤疫苗的分类方法有很多种,依据治疗原理可以划分为预防性和治疗性疫苗两大类,治疗性肿瘤疫苗还可以依据靶点类型和疫苗成药载体的不同进行划分。
5.其中以mrna作为载体的治疗性肿瘤疫苗有以下几点突出优势:(1)mrna可以同时编码多种抗原,具有mhci和mhcii结合表位的完整蛋白质,以促进体液和细胞适应性免疫反应,提供更强化的抗肿瘤免疫力。(2)与dna疫苗相比,mrna疫苗是非整合的,高度可降解的,没有插入诱变潜力。(3)与蛋白质或细胞介导的疫苗相比,mrna的ivt产生不含细胞和致病性病毒成分,没有感染可能性,正在进行临床试验测试的大多数mrna疫苗通常具有良好的耐受性,罕有注射部位反应。(4)mrna癌症疫苗的另一个优点是快速和可扩展的制造。
6.随着两种用于预防covid
‑
19的mrna
‑
lnp疫苗获得批准,mrna技术路线的可行性和优势已经得到了广泛的认可,并且随着资本的关注以及越来越多的研究人员的参与,mrna疫苗乃至mrna药物开发正在经历相当大的爆发式发展。其中一个关键的核心技术点,就是预测mrna疫苗的核心靶标:新生抗原(neoantigen)的肿瘤特异性抗原tsa。neoantigen来源于肿瘤细胞中的随机体细胞突变,不存在于正常细胞中。neoantigen可被宿主免疫系统识别为“非自身”的序列,引发强烈的免疫反应。预测个性化hla新生抗原(neoantigen)疫苗的主要步骤如下:
7.(1)鉴定和确认在患者肿瘤中表达的特异性免疫原性非同义体细胞突变。对肿瘤组织进行活组织检查以进行全外显子组或转录组测序。可以通过比较肿瘤和匹配的健康组织的序列来鉴定肿瘤的非同义体细胞突变,例如点突变和插入缺失。
8.(2)使用主要组织相容性复合物(mhc)i和ii类表位预测算法,分析和鉴定具有最高免疫原性的突变。
9.(3)基于体外结合测定结果进一步证实候选抗原的排序列表。
10.步骤(2)中,为了准确预测新生抗原免疫原性,需要知道1)哪些肽段会与mhc结合2)哪些phla能够引起免疫反应。当前已经开发了大量的hla
‑
肽段结合预测工具来预测哪些肽段将与特定的nhc进行结合。然而,仅凭mhc结合预测不足以推断免疫原性,因为此类工具无法预测哪些肽将触发t细胞反应。当前neoantigen疫苗开发的难点之一是为什么某些被感染或癌症特异性表达的并且被hla
‑
i呈递到细胞表面的肽段能给被cd8 t细胞识别并引发特定的免疫反应,而有些则不能。随着人工智能技术在生物信息学中的广泛应用,已经有领域内学者开始尝试利用数据驱动的机器学习方法,填补这一空缺。
11.其中代表性的工作与技术有,韩国延世大学团队的工作(参考文献:kim s,kim hs,kim e,et al.neopepsee:accurate genome
‑
level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information.ann oncol.2018;29(4):1030
‑
1036.doi:10.1093/annonc/mdy022)基于14个独立特征开发的机器学习算法来预测肽段免疫原性。拉霍拉学院团队的工作(参考文献:vita r,mahajan s,overton ja,et al.the immune epitope database(iedb):2018update.nucleic acids res.2019;47(d1):d339
‑
d343.doi:10.1093/nar/gky1006)通过考虑kullback
‑
leibler散度和氨基酸偏好的位置加权计算模式来预测肽段免疫原性。浙江大学团队的工作(参考文献:wu j,wang w,zhang j,et al.deephlapan:a deep learning approach for neoantigen prediction considering both hla
‑
peptide binding and immunogenicity.front immunol.2019;10:2559.published 2019nov 1.doi:10.3389/fimmu.2019.02559)基于iedb数据,采用深度学习算法,来预测肽段免疫原性。
12.以上提到的现有主流phla新生抗原免疫原性预测方法,具有以下局限性:1)只考虑有限的hla等位基因个数;2)未考虑不同受试者抗原受体谱系对肽段免疫原性的影响;3)这些算法基本未考虑肽段和mhc氨基酸序列的物理和组成特征。这些方法输出的结果,可能无法完全反应触发t细胞反应的phla特征;4)较单一化的数据模态与数据模型,使其无法充分利用生物信息大数据所蕴含的多模态信息,并缺少可扩展性。
技术实现要素:
13.本发明针对背景技术中存在的问题,提出了一种基于多模态深度编码的抗原免疫原性预测方法和系统。
14.技术方案:
15.本发明首先公开了一种基于多模态深度编码的抗原免疫原性预测方法,它包括以下步骤:
16.s1、特征选择:选定与抗原免疫原性相关的特征,作为待融合特征;
17.s2、归一化处理:设置隐嵌入维度作为不同待融合特征的最终输入维度,将s1中获取的待融合特征进行变换和尺度缩放,获得标准特征;
18.s3、特征融合:将维度相同的标准特征作线性融合操作,融合后的特征向量/矩阵输入深度神经网络,进行非线性变换与融合,获得抗原免疫原性的最终特征分数;
19.s4、构建预测模型:特征融合,构建包含最终特征分数的预测模型和优化模型;
20.s5、求解优化模型,获得最优参数的预测模型;
21.s6、使用最优参数的预测模型进行抗原免疫原性预测。
22.优选的,根据s1中特征选择的不同,s2中选定相应的归一化处理方案,以获取格式、维度统一,便于融合的标准特征,具体为:
23.‑
序列特征;使用主成分分解pca对aaindex数据库中的aaindex1数据进行变换,选取变换后的12个主成分特征,为输入的序列进行编码作为序列特征的标准特征;
24.‑
互作特征:基于aaindex数据库中的aaindex3矩阵,进行序列比对,获取比对分数,通过尺度缩放获取互作特征的标准特征;
25.‑
物理特征:表示序列的电荷、疏水性、不稳定性特征,通过尺度缩放获得物理特征的标准特征;
26.‑
组分特征:表示序列氨基酸组分特征,统计其标准氨基酸编码出现的次数作为组分特征的标准特征。
27.具体的,s1中待融合特征选择为:肽段序列特征、hla
‑
i序列特征、抗原受体
‑
肽段互作特征、肽段物理特征、肽段氨基酸组分特征。
28.具体的,s2中:
29.肽段序列特征通过以下方法获得标准特征:采用主成分分解pca对aaindex数据库中的aaindex1特征进行变换,选取变换后的12个主成分特征,对肽段的蛋白组成氨基酸进行编码作为肽段序列特征;
30.hla
‑
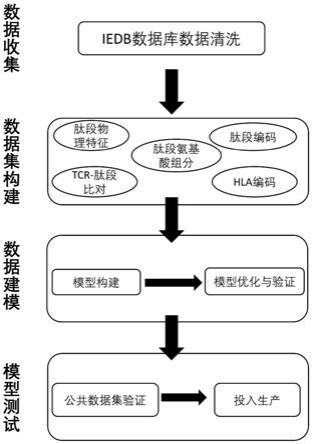
i序列特征通过以下方法获得标准特征:采用主成分分解pca对aaindex数据库中的aaindex1特征进行变换,选取变换后的12个主成分特征,对hla
‑
i序列的蛋白组成氨基酸进行编码作为hla
‑
i序列特征;
31.抗原受体
‑
肽段互作特征通过以下方法获得标准特征:基于aaindex数据库中的aaindex3特征,进行序列比对,获取比对分数,通过尺度缩放获得标准化特征,作为抗原受体
‑
肽段互作特征;
32.肽段物理特征通过尺度缩放获得标准特征,以保证模型训练优化过程的数值稳定性;
33.肽段氨基酸组分特征通过以下方法获得标准特征:统计肽段中标准氨基酸编码出现的次数,作为肽段氨基酸组分特征。
34.具体的,抗原受体
‑
肽段互作特征的尺度变化公式为:
[0035][0036]
式中,p表示抗原受体
‑
肽段互作分数,p
′
表示作为标准特征的抗原受体
‑
肽段互作分数。
[0037]
具体的,肽段物理特征的尺度变化公式为:
[0038][0039]
式中,x表示肽段物理特征分数,x
′
表示作为标准特征的肽段物理特征分数。
[0040]
具体的,s3特征融合中,所述线性融合操作包括点乘、或加和、或组合成特征矩阵。
[0041]
具体的,s4中构建优化模型:
[0042][0043]
式中,f是含可学习参数的预测模型;w表示该模型中可学习参数,包括各融合特征获取时方案权重;x
n
表示输入的特定数据,y
n
是训练数据中免疫原性可信度数值;n表示样本总数。
[0044]
优选的,s5中求解优化模型:多次遍历所有训练数据,利用基于随机梯度优化方法的优化器进行优化,得到最优的模型参数,获得预测模型f
w
。
[0045]
优选的,采用beta分布,对iedb数据库中具有实验验证的免疫原性结果进行编码,转换为回归拟合问题,基于此来提高训练模型可靠性;所述训练数据经过包括重抽样、剔除负样本的数据处理,避免使用的训练的数据正负样本量不平衡的问题。
[0046]
本发明还公开了一种基于多模态深度编码的新抗原免疫原性预测系统,它包括:
[0047]
‑
数据收集模块,整理iedb数据库中具有免疫原性验证结果的肽段及其mhc
‑
i配体数据对;
[0048]
‑
数据集构建,根据整理的iedb数据库中具有免疫原性验证结果的肽段及其mhc
‑
i配体数据对,构建正负样本。
[0049]
‑
数据建模模块,构建预测模型并求解预测模型。
[0050]
优选的,所述数据建模模块包括:
[0051]
‑
特征选择模块,选定与肽段免疫原性相关的特征,作为待融合特征;
[0052]
‑
归一化处理模块,将待融合特征进行变换和尺度缩放,获得标准特征;
[0053]
‑
特征融合模块,将多模态的标准特征输入深度神经网络进行融合,得到肽段免疫原性最终特征分数;
[0054]
‑
预测模型构建模块,构建包含最终特征分数的预测模型和优化模型;
[0055]
‑
预测模型求解模块,计算获得最优参数的预测模型。
[0056]
优选的,所述特征融合模块中,采用可变长的维度的输入设计,以便未来加入更多新模态特征的接口。
[0057]
优选的,采用beta分布,对iedb数据库中具有实验验证的免疫原性结果进行编码,转换为回归拟合问题,基于此来提高训练模型可靠性;所述训练数据经过包括重抽样、剔除负样本的数据处理,避免使用的训练的数据正负样本量不平衡的问题。
[0058]
更优的,它还包括:
[0059]
‑
测试模块,整理包含在文献中但不包含在数据集构建模块出现的数据,以待优化得到最优模型后,验证测试模型的对于未曾见过的免疫原性数据对的预测效果。
[0060]
本发明的有益效果
[0061]
本发明提出了一种基于多模态深度编码的抗原免疫原性预测方法和系统,包括:1)基于抗体结合部位信息,包含更多hla等位基因个数;2)可变长肽段编码方式,涵盖主要hla结合肽段长度;3)考虑抗原受体谱系对肽段免疫原性的影响;4)包含肽段和mhc序列的物理和氨基酸组成特征;5)采用beta分布,对iedb数据库中具有实验验证的免疫原性结果进行编码,转换为回归拟合问题,基于此来提高训练模型可靠性;6)基于归一化处理,最终能得到格式、维度统一,便于融合的特征向量;7)基于可变长的维度的输入设计,以便未来
加入更多新模态特征的接口。未来加入的新特征只要是能被现有机器学习方法进行编码的,理论上没有任何限制,这也是我们模型“可拓展性”优点的体现。
附图说明
[0062]
图1多模态深度编码的抗原免疫原性预测方法计算流程图
[0063]
图2多模态深度编码的抗原免疫原性预测系统总结构图
[0064]
图3为10折交叉验证auc评估结果图
[0065]
图4为10折交叉验证auc
‑
pr评估结果图
具体实施方式
[0066]
下面结合实施例对本发明作进一步说明,但本发明的保护范围不限于此:
[0067]
如图1所示,本发明提出的多模态深度编码的抗原免疫原性预测系统分为四部分,下面针对数据收集,数据集构建,模型建构与优化和模型测试进行详细阐述。
[0068]
(a)数据收集
[0069]
该模块为根据iedb数据库(参考文献:vita r,mahajan s,overton ja,et al.the immune epitope database(iedb):2018update.nucleic acids res.2019;47(d1):d339
‑
d343.doi:10.1093/nar/gky1006)公开资源,整理与hla
‑
i免疫原性有关结果,具体包括:
[0070]
i.选定t
‑
cellassay中的linearpeptide;
[0071]
ii.选定host organism为homo sapiens;
[0072]
iii.选定拥有完整hla等位基因命名的hla
‑
i等位基因型;
[0073]
iv.选取肽段长度为9或者10;
[0074]
v.选定具有qualitativemeasure信息的数据行;过滤缺失numberofsubjectstested或numberofsubjectsresponded数值的数据行;过滤重复及结论冲突的数据行;
[0075]
(b)数据集构建
[0076]
基于(a)中的方法收集hla
‑
i免疫原性的正负样本,构建模型训练数据集,具体如下:
[0077]
i.使用抗体结合部位序列,代表hla
‑
i序列;
[0078]
ii.采用主成分分解pca,对aaindex数据库中的aaindex1特征进行变换,选取变换后的12个主成分特征,对肽段和hla中组分氨基酸进行编码;
[0079]
iii.使用开源计算r包peptides(参考文献:osorio d,rondon
‑
villarreal p,torres r(2015).“peptides:a package for data mining of antimicrobial peptides.”the r journal,7(1),4
‑
14.issn2073
‑
4859.),计算肽段的序列的电荷、疏水性、不稳定性物理特征,并进行尺度缩放;
[0080]
iv.基于aaindex数据库中的aaindex3特征,将肽段与公开人类tcr数据集进行序列比对,获取比对分数;获取每一个肽段对应比对值分数的平均值,并进行尺度缩放;
[0081]
v.统计每一个肽段对应的标准氨基酸编码个数。
[0082]
vi.基于(a)中的方法收集hla
‑
i免疫原性的正负样本中,qualitativemeasure取值为以下五种类型:negative,positive,positive
‑
high,positive
‑
intermediate,
positive
‑
low,分别对应:无免疫原性;有免疫原性;强免疫原性;中等免疫原性及弱免疫原性,基于其对应的number of subiects tested和number of subjects responded信息,使用如下beta分布,生成10000个随机数,取这些值的均值作为免疫原性分数,进行免疫原性编码:
[0083][0084]
其中,t表示number of subjects tested值;s表示number of subjects responded值。以immu
score
值表征每行数据免疫原性程度。
[0085]
选取以下文章中免疫原性结果,构建公共验证数据集,公共验证数据集数据集hla及肽段编码方式同模型训练数据集:
[0086]
1)tesla数据集:wells dk,van buuren mm,dang kk,et al.key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction.cell.2020;183(3):818
‑
834.e13.doi:10.1016/j.cell.2020.09.015
[0087]
2)emma数据集:jappe ec,garde c,ramarathinam sh,et al.thermostability profiling of mhc
‑
bound peptides:a new dimension in immunopeptidomics and aid for immunotherapy design.nat commun.2020;11(1):6305.published 2020 dec 9.doi:10.1038/s41467
‑
020
‑
20166
‑4[0088]
3)ott数据集:ott pa,hu z,keskin db,et a1.an immunogenic personal neoantigen vaccine for patients with melanoma[published correction appears in nature.2018 mar14;555(7696):402].nature.2017;547(7662):217
‑
221.doi:10.1038/nature22991
[0089]
4)bulik
‑
sullivan数据集:bulik
‑
sullivan,b.,busby,j.,palmer,c.et a1.deep learningusing tumor hla peptide mass spectrometry datasets improves neoantigen identification.nat biotechnol 37,55
‑
63(2019).doi.org/10.1038/nbt.4313
[0090]
5)robbins数据集:robbins pf,lu yc,el
‑
gamil m,et al.mining exomic sequencing data to identifymutated antigens recognized by adoptively transferred tumor
‑
reactive t cells.nat med.2013;19(6):747
‑
752.doi:10.1038/nm.3161
[0091]
(c)基于深度编码和多模态数据的模型建构与优化
[0092]
如图2的计算流程图所示,我们对模块(b)中的数据集进行编码、并建立模型。具体实施如下:
[0093]
i.选择k
‑
折交叉验证(k
‑
fold cross validatio)的统计学方法构建模型训练、测试数据集。
[0094]
ii.构建深度学习模型,模型结构如图2。将肽段氨基酸组成、肽段物理特征、tcr
‑
肽段比对、编码肽段和编码hla输入特征融合层,获得如下优化模型:
[0095][0096]
其中,f是含可学习参数的预测模型;w表示该模型中可学习参数,具体包括循环编码肽段;x
n
表示输入的特定数据,y
n
是训练数据中免疫原性可信度数值,既immu
score
值;n表示样本总数。
[0097]
iii.最优模型的求解,采用批次随机梯度下降策略(参考文献:goyal,priya,et al.
″
accurate.large minibatch sgd:training imagenet in 1 hour.
″
arxiv preprint arxiv:1706.02677(2017).):在多个轮次中,将训练数据分批次输入模型,计算如上的损失函数与梯度,并利用梯度下降更新模型。具体来说,我们采用adma优化器(参考文献:kingma,diederik p.,and jimmy ba.
″
adam:a method for stochastic optimization.
″
arxiv preprint arxiv:1412.6980(2014).),其用一阶梯度估计高阶梯度,并能自动调节优化的步长,是模型优化过程更加稳定与稳健。
[0098]
(d)模型测试与机器学习评价指标
[0099]
i.模型评估
[0100]
评估和交叉验证是测量模型性能的标准方法。它们都生成可检查或与其他模型比较的评估指标。
[0101]
我们采用接收者操作特征曲线(receiver operating characteristic curve,简称roc曲线)下面积auc与准确度
‑
召回率(precision
‑
recall,简称pr)曲线下面积auc
‑
pr来评价优化后模型的预测能力与性能(表1):
[0102]
表1预测模型评价指标
[0103]
评价指标描述aucroc曲线下而积auc
‑
prpr曲线下面积
[0104]
此处,采用10折交叉验证,将模型训练数据集拆分为10部分,其中一个部分保留用于测试,其他9部分用于训练。此过程重复10次。
[0105]
如图3为10折交叉验证auc评估结果,如图4为10折交叉验证auc
‑
pr评估结果。可以看到,模型在每一折上都具有很高的auc值和auc
‑
pr值,均值分别为0.82和0.8。证明模型具有很好的泛化能力,能够很好的应对实际生产研发中的预测问题。
[0106]
ii.模型对比
[0107]
1.基于真实数据集tesla数据集,将模型与两个广泛使用的免疫原性预测模型iedb和deephlapan进行比较。评价指标设定如下(表2):以0.5为阈值,计算灵敏度与精准度ppv,评估结果见表3。根据打分结果降序排列,我们分别选取了前20(top20)及前50(top50)计算ppv,可见本模型immu
‑
d的结果是三种方法中最优的。基于全部数据,计算灵敏度,本模型immu
‑
d也是最高的,两倍于deephlapan的结果。
[0108]
表2模型比较评估指标
‑1[0109]
评价指标描述灵敏度真阳性/(真阳性 假阴性)
精准度/ppv真阳性/(真阳性 假阳性)
[0110]
表3模型比较评估结果
‑1[0111][0112][0113]
2.基于真实数据集emma数据集,将模型与其文中提到的模型stabilitypredictor,mixmhcpred,netmhcpan
‑
4.0(el),netmhcpan
‑
4.0(ba),mhcflurry进行比较。评价指标设定如下(表4):以0.5为阈值,计算auc与精准度ppv,评估结果见表3。根据打分结果降序排列,我们分别选取了前10(top10)计算ppv。结果表明(表5),在auc基本持平的情况下,本模型immu
‑
d与基于复杂实验结果构建的模型stabilitypredictor具有相同的ppv值,远优于其余四个模型结果。
[0114]
表4模型比较评估指标
‑2[0115]
评价指标描述aucroc曲线下面积精准度/ppv真阳性/(真阳性 假阳性)
[0116]
表5模型比较评估结果
‑2[0117] ppv
‑
topl0aucstability predictor0.90.75mixmhcpred0.70.7netmhcpan
‑
4.0(el)0.60.68netmhcpan
‑
4.0(ba)0.60.67mhcflurry0.70.65immu
‑
d0.90.66
[0118]
iii.真实数据结果展示
[0119]
真实生产环境下,由于相关的限制,通常只会对部分候选肽段进行后续实验验证。为了验证模型在实际生产中的作用,我们以精准度ppv为判别指标,分别计算模型在前10(top10)、前20(top20)、前30(top30)及整体数据上的表现,结果见表6。从结果可知,即使在高阳性
‑
阴性比数据上,我们的模型也能很好的捕获阳性结果。其也佐证了我们模型在真实生产环境下的价值。
[0120]
表6真实数据结果
[0121]
[0122]
应当理解的是,本发明的应用不限于上述的据力。对本领域从业技术人员来说,可以根据上述说明加以改进或者变换,特别是基本模型选取、免疫指标构建方法及相关特征值的添加。所有这些改进和变换,以及参数相关的调节和选取,都应属于本发明所附权利要求的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。