14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法
技术领域
1.本发明涉及计算机辅助药物分子设计技术领域,特别涉及一种14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法。
背景技术:
2.14
‑3‑
3蛋白是一个酸性蛋白家族的统称,成员之间具有高度的序列保守性和结构相似性,蛋白质的分子量在30kda左右。这些蛋白存在于多种不同类型的真核细胞和组织中,在体内以同源或异源二聚体的形式存在。形成二聚体后在蛋白结构上呈现出一个凹槽状结构域,该结构域中带正电荷的残基(如k49、r56和r127)能够与带有磷酸化丝氨酸/苏氨酸(ps/t)位点的配体结合并发生相互作用。因此,配体磷酸化是可以与14
‑3‑
3蛋白发生相互作用的必要条件。14
‑3‑
3蛋白质和特定的磷酸化配体结合之后会形成特定的复合体,复合体能够激活和参与一些细胞内的信号转导过程。研究证实,14
‑3‑
3家族大部分成员都能参与这些过程。通过与特定配体结合,14
‑3‑
3蛋白能够参与控制不同的细胞活动,包括代谢、转录、细胞周期、分化、迁移和凋亡等。此外,14
‑3‑
3蛋白也是产生癌细胞药物抗性的一个关键因子。很多经研究确认的14
‑3‑
3蛋白结合配体是原癌基因或癌基因的表达产物,例如c
‑
raf
‑
1,pymt,bcr/ab1,htert,igf
‑
ir,β
‑
catenin。除了与癌症有关的配体发生结合外,研究发现有一些14
‑3‑
3亚型在肿瘤细胞中的基因转录水平升高,在癌症化疗后复发或预后不良的癌症患者病例中也发现了14
‑3‑
3蛋白基因的过量表达。这些结果都表明,14
‑3‑
3作为关键信号因子参与了多种人类癌症化疗抗性的产生,14
‑3‑
3的过度表达与患者的不良预后情况有关。这使得14
‑3‑
3有可能成为癌症诊断的新型生物标志物,也是一些癌症诊疗的有效候选靶点之一。在人源14
‑3‑
3家族中已经发现有七种结构相似但表现不同的亚型,分别用希腊字母命名为β,γ,ε,τ,σ,η。每个亚型对应了一个特定的编码基因。在这些亚型的蛋白多序列比对中发现存在五个同源区块。通过比较整体的蛋白晶体结构,发现14
‑3‑
3亚型之间呈现出高度的结构相似性。由于在序列和结构上的高度保守,亚型之间在某些功能上能够互补。一些特殊情况下,一种亚型的功能缺陷可以由另一种亚型来补偿。虽然如此,每个14
‑3‑
3亚型的二聚体偏好性和配体结合特异性上存在明显的差异。在某些特定组织细胞中发现,在二聚体形式上有的14
‑3‑
3亚型倾向于形成同源二聚体,有的倾向于形成异源二聚体。14
‑3‑
3ε倾向于与β、γ和ζ形成异源二聚体,且没有发现同源二聚体。14
‑3‑
3σ主要是同源二聚体的形式,γ则发现同时具有两种二聚体形式。对于特定的14
‑3‑
3亚型,其能够结合的配体也不同。与14
‑3‑
3蛋白结合的配体主要是磷酸化形式,早期的研究发现ps/t及其周围区域都会参与14
‑3‑
3和配体的结合。所以14
‑3‑
3蛋白的配体结合位点包含了ps/t及其周边区域的特定序列模式。目前在14
‑3‑
3s结合位点上存在着三种主要序列模式:rsxpsxp、pxxpsxp和位于配体羧基末端的swptx。除此之外,还有几种例外情况,例如结合位点无磷酸化或与前述模式不同。这些亚型/亚型和亚型/配体之间的特异性可能源自于14
‑3‑
3亚型之间的结构和序列的轻微差异造成的。亚型之间在结构上的差异可能引起亚型与不同序列排列的磷酸化多肽之间发生相互作用的亲和力的差异。这种差异可以帮助更多地
了解14
‑3‑
3蛋白与配体识别和结合的细节,从而更有效地进行以14
‑3‑
3为靶点的药物设计。14
‑3‑
3蛋白与磷酸化多肽的亲和力可以通过高通量的多肽文库获得。但是仍有很多限制因素存在。例如对于三肽来说,覆盖20个氨基酸的所有序列排列组合的数目是8000个。
技术实现要素:
3.本发明的主要目的在于提供14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法,可以有效解决背景技术中的问题。
4.为实现上述目的,本发明采取的技术方案为:
5.一种14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法,包括以下步骤:
6.步骤s1收集和处理数据,收集不同来源的所有七种亚型与三肽配体的亲和力数据,并对数据集进行预处理和质量检查;
7.步骤s2构建14
‑3‑
3配体亲和力的定量构效关系模型,用来自dpps的氨基酸描述符代表每个磷酸化多肽上的理化特征并作为预测变量,对应14
‑3‑
3每个亚型的配体亲和力作为响应变量,用惩罚回归方法建模;
8.步骤s3预测14
‑3‑
3蛋白与配体的亲和力,利用依据14
‑3‑
3多肽文库数据构建的定量构效关系模型预测所有可能序列组合的磷酸化三肽配体与14
‑3‑
3亚型的相对亲和力。
9.进一步地,还包括以下步骤:
10.步骤s4配体亲和力的聚类分析,将标准化的14
‑3‑
3配体亲和力数据,利用了二维层次聚类方法进行分析。
11.进一步地,所述步骤s1中,所述对数据集进行预处理和质量检查为:
12.提高整体数据的辨识度,对原始的荧光值进行了以2为底数的对数转换;
13.将数值相对小于0的序列从数据集中删除。
14.进一步地,所述步骤s2还包括:
15.数据集中80%的亲和力数据作为训练数据,剩下的作为测试数据;
16.根据每个定量构效关系模型中配体位点上不同物化性质的系数,评估多肽上每个位点的物化性质对14
‑3‑
3配体亲和力的贡献,确定14
‑3‑
3配体结合中的关键位点和关键氨基酸;
17.对每个定量构效关系模型的预测准确性进行评估,用训练得到的模型重新对整个数据集进行预测,每个磷酸化多肽配体获得一个预测值,从预测结果中筛选出高亲和力的磷酸化多肽并与原始实验数据进行比较。
18.进一步地,所述步骤s2还包括:
19.对于每个14
‑3‑
3配体序列,它与14
‑3‑
3的亲和力值f(x)由包含p个变量的向量表示:
20.f(x)=β0 β1x1 β2x2 β3x3
…
β
p
x
p
21.其中β0是模型常数,x1,x2,x3...xp是一系列的氨基酸描述符,β1,β2,β3...βp是基于回归模型得到的相应的系统,p为常数;
22.使用线性回归构建序列与亲和力相关的定量构效关系模型;
23.所述惩罚回归方法为elastic net方法:
[0024][0025]
采用10
‑
fold交叉验证的方法来优化训练集其中的参数λ和α,使交叉验证的误差最小,训练集随机分为10个部分,然后每个部分都用作模型在其他部分的测试,然后选择λ和α的最优值作为模型参数。
[0026]
进一步地,所述步骤s2还包括:
[0027]
同时计算预测与真实数据集之间的r平方和rmse,以评估模型拟合度,rmse的计算方式为:
[0028][0029]
ei是实验数据集中第i个亲和力值,pi是来自模型的第i个预测结果;
[0030]
r的平方的计算方式为:
[0031][0032]
e是实验数据集中14
‑3‑
3配体的平均亲和力。
[0033]
进一步地,所述步骤s3还包括:
[0034]
预测得到的数据依据不同亚型进行数据标准化处理,使其平均值为0,方差为1,标准化后的数据视为14
‑3‑
3蛋白与配体亲和力的预测结果。
[0035]
进一步地,所述步骤s4还包括:
[0036]
在聚类前对亲和力数值在亚型间进行标准化,通过欧氏距离方法获得距离矩阵,使用completeagglomeration方法进行聚类。
[0037]
进一步地,所述步骤s4还包括:通过r软件heatmap.2包中的gplots函数实现并进行可视化,配体多肽序列的保守性分析通过将多肽序列上传到weblogo网络服务器进行。
[0038]
与现有技术相比,本发明具有如下有益效果:
[0039]
从数据集中提取多肽序列和配体相对亲和力数据,利用一种氨基酸描述符dpps来生成训练和测试数据集,利用定量构效关系方法建立了一种有效的亲和力预测模型;
[0040]
通过使用elastic net方法解决了ridge和lasso的一些限制,且elastic net模型具有更好的拟合能力。
附图说明
[0041]
图1为本发明的14
‑3‑
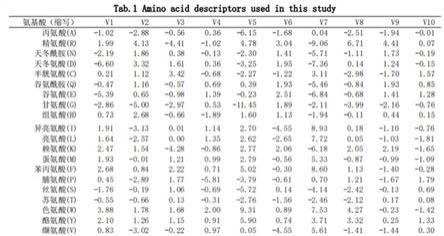
3蛋白质与磷酸化配体相对亲和力的估计和预测方法的的氨基酸描述符表。
[0042]
图2为本发明的14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法的比较散点图。
[0043]
图3为本发明的14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法的配体位点上的物化性质对亲和力的贡献示意图。
[0044]
图4为本发明的14
‑3‑
3蛋白质与磷酸化配体相对亲和力的配体序列和14
‑3‑
3蛋白
有直接的相互作用关系表。
[0045]
图4为本发明的14
‑3‑
3蛋白质与磷酸化配体相对亲和力的预测出的所有磷酸化配体亲和力的层次聚类分析结果示意图。
具体实施方式
[0046]
为使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施方式,进一步阐述本发明。
[0047]
如图1
‑
5所示的一种14
‑3‑
3蛋白质与磷酸化配体相对亲和力的估计和预测方法,包括以下步骤:
[0048]
步骤s1收集和处理数据,收集不同来源的所有七种亚型与三肽配体的亲和力数据,并对数据集进行预处理和质量检查;
[0049]
步骤s2构建14
‑3‑
3配体亲和力的定量构效关系模型,用来自dpps的氨基酸描述符代表每个磷酸化多肽上的理化特征并作为预测变量,对应14
‑3‑
3每个亚型的配体亲和力作为响应变量,用惩罚回归方法建模;
[0050]
步骤s3预测14
‑3‑
3蛋白与配体的亲和力,利用依据14
‑3‑
3多肽文库数据构建的定量构效关系模型预测所有可能序列组合的磷酸化三肽配体与14
‑3‑
3亚型的相对亲和力。
[0051]
其中,还包括以下步骤:
[0052]
步骤s4配体亲和力的聚类分析,将标准化的14
‑3‑
3配体亲和力数据,利用了二维层次聚类方法进行分析。
[0053]
其中,所述步骤s1中,所述对数据集进行预处理和质量检查为:
[0054]
提高整体数据的辨识度,对原始的荧光值进行了以2为底数的对数转换;
[0055]
将数值相对小于0的序列从数据集中删除。
[0056]
其中,所述步骤s2还包括:
[0057]
数据集中80%的亲和力数据作为训练数据,剩下的作为测试数据;
[0058]
根据每个定量构效关系模型中配体位点上不同物化性质的系数,评估多肽上每个位点的物化性质对14
‑3‑
3配体亲和力的贡献,确定14
‑3‑
3配体结合中的关键位点和关键氨基酸;
[0059]
对每个定量构效关系模型的预测准确性进行评估,用训练得到的模型重新对整个数据集进行预测,每个磷酸化多肽配体获得一个预测值,从预测结果中筛选出高亲和力的磷酸化多肽并与原始实验数据进行比较。
[0060]
其中,所述步骤s2还包括:
[0061]
对于每个14
‑3‑
3配体序列,它与14
‑3‑
3的亲和力值f(x)由包含p个变量的向量表示:
[0062]
f(x)=β0 β1x1 β2x2 β3x3
…
β
p
x
p
[0063]
其中β0是模型常数,x1,x2,x3...xp是一系列的氨基酸描述符,β1,β2,β3...βp是基于回归模型得到的相应的系统,p为常数;
[0064]
使用线性回归构建序列与亲和力相关的定量构效关系模型;
[0065]
所述惩罚回归方法为elastic net方法:
[0066][0067]
采用10
‑
fold交叉验证的方法来优化训练集其中的参数λ和α,使交叉验证的误差最小,训练集随机分为10个部分,然后每个部分都用作模型在其他部分的测试,然后选择λ和α的最优值作为模型参数。
[0068]
其中,所述步骤s2还包括:
[0069]
同时计算预测与真实数据集之间的r平方和rmse,以评估模型拟合度,rmse的计算方式为:
[0070][0071]
ei是实验数据集中第i个亲和力值,pi是来自模型的第i个预测结果;
[0072]
r的平方的计算方式为:
[0073][0074]
e是实验数据集中14
‑3‑
3配体的平均亲和力。
[0075]
其中,所述步骤s3还包括:
[0076]
预测得到的数据依据不同亚型进行数据标准化处理,使其平均值为0,方差为1,标准化后的数据视为14
‑3‑
3蛋白与配体亲和力的预测结果。
[0077]
其中,所述步骤s4还包括:
[0078]
在聚类前对亲和力数值在亚型间进行标准化,通过欧氏距离方法获得距离矩阵,使用completeagglomeration方法进行聚类。
[0079]
其中,所述步骤s4还包括:通过r软件heatmap.2包中的gplots函数实现并进行可视化,配体多肽序列的保守性分析通过将多肽序列上传到weblogo网络服务器进行。
[0080]
14
‑3‑
3蛋白是一个酸性蛋白家族的统称,成员之间具有高度的序列保守性和结构相似性,蛋白质的分子量在30kda左右。这些蛋白存在于多种不同类型的真核细胞和组织中,在体内以同源或异源二聚体的形式存在。形成二聚体后在蛋白结构上呈现出一个凹槽状结构域,该结构域中带正电荷的残基(如k49、r56和r127)能够与带有磷酸化丝氨酸/苏氨酸(ps/t)位点的配体结合并发生相互作用。因此,配体磷酸化是可以与14
‑3‑
3蛋白发生相互作用的必要条件。14
‑3‑
3蛋白质和特定的磷酸化配体结合之后会形成特定的复合体,复合体能够激活和参与一些细胞内的信号转导过程。研究证实,14
‑3‑
3家族大部分成员都能参与这些过程。通过与特定配体结合,14
‑3‑
3蛋白能够参与控制不同的细胞活动,包括代谢、转录、细胞周期、分化、迁移和凋亡等。此外,14
‑3‑
3蛋白也是产生癌细胞药物抗性的一个关键因子。很多经研究确认的14
‑3‑
3蛋白结合配体是原癌基因或癌基因的表达产物,例如c
‑
raf
‑
1,pymt,bcr/ab1,htert,igf
‑
ir,β
‑
catenin。除了与癌症有关的配体发生结合外,研究发现有一些14
‑3‑
3亚型在肿瘤细胞中的基因转录水平升高,在癌症化疗后复发或预后不良的癌症患者病例中也发现了14
‑3‑
3蛋白基因的过量表达。这些结果都表明,14
‑3‑
3作为关键信号因子参与了多种人类癌症化疗抗性的产生,14
‑3‑
3的过度表达与患者的不良预
后情况有关。这使得14
‑3‑
3有可能成为癌症诊断的新型生物标志物,也是一些癌症诊疗的有效候选靶点之一。在人源14
‑3‑
3家族中已经发现有七种结构相似但表现不同的亚型,分别用希腊字母命名为β,γ,ε,τ,σ,η。每个亚型对应了一个特定的编码基因。在这些亚型的蛋白多序列比对中发现存在五个同源区块。通过比较整体的蛋白晶体结构,发现14
‑3‑
3亚型之间呈现出高度的结构相似性。由于在序列和结构上的高度保守,亚型之间在某些功能上能够互补。一些特殊情况下,一种亚型的功能缺陷可以由另一种亚型来补偿。虽然如此,每个14
‑3‑
3亚型的二聚体偏好性和配体结合特异性上存在明显的差异。在某些特定组织细胞中发现,在二聚体形式上有的14
‑3‑
3亚型倾向于形成同源二聚体,有的倾向于形成异源二聚体。14
‑3‑
3ε倾向于与β、γ和ζ形成异源二聚体,且没有发现同源二聚体。14
‑3‑
3σ主要是同源二聚体的形式,γ则发现同时具有两种二聚体形式。对于特定的14
‑3‑
3亚型,其能够结合的配体也不同。与14
‑3‑
3蛋白结合的配体主要是磷酸化形式,早期的研究发现ps/t及其周围区域都会参与14
‑3‑
3和配体的结合。所以14
‑3‑
3蛋白的配体结合位点包含了ps/t及其周边区域的特定序列模式。目前在14
‑3‑
3s结合位点上存在着三种主要序列模式:rsxpsxp、pxxpsxp和位于配体羧基末端的swptx。除此之外,还有几种例外情况,例如结合位点无磷酸化或与前述模式不同。这些亚型/亚型和亚型/配体之间的特异性可能源自于14
‑3‑
3亚型之间的结构和序列的轻微差异造成的。亚型之间在结构上的差异可能引起亚型与不同序列排列的磷酸化多肽之间发生相互作用的亲和力的差异。这种差异可以帮助更多地了解14
‑3‑
3蛋白与配体识别和结合的细节,从而更有效地进行以14
‑3‑
3为靶点的药物设计。14
‑3‑
3蛋白与磷酸化多肽的亲和力可以通过高通量的多肽文库获得。但是这种实验仍有很多限制因素存在。例如对于三肽来说,覆盖20个氨基酸的所有序列排列组合的数目是8000个。由于这种排列方式数量大,所以有必要开发开发一种计算方法来基于现有的数据集预测未知磷酸化多肽配体与14
‑3‑
3的亲和力。在计算方法层面上,基于定量构效关系的建模方法已被广泛用于推断分子功能和寻找具有高活性的化合物,如亲和力、毒性和抗菌活性等。定量构效关系建模方法可以基于氨基酸序列进行多肽生物活性的预测。具体来说,多肽文库中磷酸化配体与14
‑3‑
3蛋白的亲和力可以被看作是多肽序列的一种生物活性。多肽中氨基酸的理化性质作为预测变量,亲和力作为反应变量,通过统计方法联系起来建立预测性定量构效关系模型。在统计建模中,回归的方法如最小二乘回归(pls)、多元线性回归(mlr)、主成分回归(pcr)和支持向量机(svm)都可以用于建立模型,线性回归模型更简单、准确和易于解释。通过氨基酸描述符来代表多肽序列理化特性,这在蛋白相互作用的机制是合理的。因此,这里选择线性回归方法进行建模。基于生物数据建立14
‑3‑
3蛋白配体亲和力的预测性定量构效关系模型,并大规模预测14
‑3‑
3亚型和配体之间的亲和力,可以帮助更好的理解14
‑3‑
3与配体之间的关系,特别是亚型之间的差异。在本技术中详细介绍了基于已经发表的数据集构建用于确定磷酸化多肽与14
‑3‑
3亲和力的预测模型的方法。从数据集中提取多肽序列和配体相对亲和力数据,利用一种氨基酸描述符dpps来生成训练和测试数据集,利用定量构效关系方法建立了一种有效的亲和力预测模型,预测结果与其他研究中已经发现的配体序列模式进行交叉验证。在此基础上,利用聚类方法鉴定了14
‑3‑
3亚型之间的配体结合特异性,并发现了一些特定的序列模式。
[0081]
所使用数据集是从已经公开发表的14
‑3‑
3多肽文库中下载和提取的,其中包含不同来源的所有七种亚型与三肽配体的亲和力。该文库中的亲和力数据由包含了1000个磷酸
化多肽的微阵列芯片产生,并分别检测了七个14
‑3‑
3亚型。磷酸化多肽根据位点被分成两个子库:氨基端库(n库)p
‑
3p
‑
2p
‑1‑
ps
‑
x 1x 2x 3和羧基端库(c库)x
‑
3x
‑
2x
‑1‑
ps
‑
p 1p 2p 3各500个序列。p和x代表不同氨基酸残基,下标代表残基与ps的相对位置,如果位于ps的氨基端为负数。其中p
±
1/2包含了十种代表性氨基酸(ala,glu,phe,gly,lys,leu,pro,gln,arg,val),p
±
3包含了其中五种氨基酸(glu,phe,leu,pro,arg)。而x代表包含其他氨基酸的混合物。芯片的荧光值是由cy3标记的14
‑3‑
3蛋白上的荧光信号产生,所以代表了磷酸化多肽和14
‑3‑
3亚型之间的相对亲和力。
[0082]
数据预处理由于原始数据是由荧光信号产生,所以整体数据的分布不均衡。在建立模型之前,所有的数据都需要经过预处理,并进行质量检查。首先,为了提高整体数据的辨识度,在分析前对原始的荧光值进行了以2为底数的对数转换(log2),处理后的数据代表了14
‑3‑
3亚型与每个磷酸化多肽的相对结合亲和力。其次,将数值相对较低(<0)的序列从数据集中删除。低相对亲和力值表明相应的多肽序列对蛋白和配体的结合没有贡献,这可能对建立模型造成负面影响。氨基酸描述符的选择和使用磷酸化多肽片段中的氨基酸被提取出来,生成预测变量矩阵用于后续模型的构建。每个多肽序列上的氨基酸残基可以通过描述符来代表。在14
‑3‑
3和配体的相互作用中,非结合效应如静电力、范德华力、疏水作用和氢键发挥关键作用。搜集了能够体现这些效应的几种氨基酸描述符,并最终确定使用dpps。dpps由10个变量组成,用于描述氨基酸的电子性、构效特性、疏水性和氢键作用,如图1,v1
‑
v4离子效应,v5
‑
v6空间效应,v7
‑
v8疏水性,v9
‑
v10氢键,第一列中的氨基酸缩写用作表示多肽中的氨基酸残基。对于数据集中的三肽序列,每个位置上氨基酸残基的物化参数由10个dpps描述符来代表,所以每个序列可以产生30个变量。根据这些变量,为七个14
‑3‑
3亚型的n/c库分别建立了定量构效关系模型。
[0083]
数据集经过预处理后被分为两部分,80%用于模型训练,剩下部分作为测试集。例如在14
‑3‑
3σ的n库中有500条数据,随机选择其中的400个作为训练集,其余100个样本作为测试集。训练集用于模型构建,建模完成后,测试数据集被用来确认性能。在定量构效关系模型中,首先假设14
‑3‑
3配体的亲和力与通过氨基酸描述符提取变量构建的预测变量x之间存在线性关系。对于每个14
‑3‑
3配体序列,它与14
‑3‑
3的亲和力值f(x)由包含p个变量的向量表示:
[0084]
f(x)=β0 β1x1 β2x2 β3x3
…
β
p
x
p
[0085]
其中β0是模型常数,x1,x2,x3...xp是一系列的氨基酸描述符,β1,β2,β3...βp是基于回归模型得到的相应的系统,p在这里为30。原始数据中n/c库中的多肽序列被氨基酸描述符转化为二维数据矩阵,那么线性回归的方法,如pls,可用于构建序列与亲和力相关的定量构效关系模型。使用惩罚回归方法包括lasso、ridge和elastic net,可以建立惩罚线性回归模型。惩罚回归通过在方程中加入一个约束值λ,以最小化残差的平方和。引入这个约束值可以将方程中一些系数减低到0,这样的话对模型贡献较少的变量的系数就可以接近或等于0。lasso和ridge回归方法的主要区别在于其中使用的惩罚项不一样。其中ridge使用l2范数,也就是系数的平方和而lasso使用l1范数,也就是系数的
net回归方法建模。数据集中80%的亲和力数据作为训练数据,剩下的作为测试数据。在测试数据中的结果清楚地显示本技术中所使用建模方法的稳健性。特别是对于14
‑3‑
3ζ,在测试数据结果中n库的总体r2和rmse值为0.790和0.619,c库则为0.744和0.704,如图2。七个亚型之间的预测效果不完全一致,这可能是因为相对结合亲和力值在肽库中的分布不同。根据每个定量构效关系模型中配体位点上不同物化性质的系数,据此评估多肽上每个位点的物化性质对14
‑3‑
3配体亲和力的贡献,以确定14
‑3‑
3配体结合中的关键位点和关键氨基酸如图3。图中每个位点物化性质的顺序与图1中的顺序保持一致,从左到右分别为v1到v10。相对于ps位点,上游的
‑
2和
‑
1位,以及下游 1位上残基对总体亲和力的贡献主要来自于氨基酸的疏水和静电相互作用。同时也发现,有些相互作用在14
‑3‑
3亚型之间并不是一致的。例如,
‑
2位的静电相互作用只在14
‑3‑
3σ、τ和ζ中发现,这些亚型可以酸/碱性氨基酸发生作用而产生高亲和力。所以arg和lys在
‑
1位置上更有优势,而非极性残基在 1的位置上更具有利于蛋白和配体的结合。同时,也对每个定量构效关系模型的预测准确性进行了评估。用训练得到的模型重新对整个数据集进行预测,每个磷酸化多肽配体获得一个预测值。从预测结果中筛选出高亲和力的磷酸化多肽并与原始实验数据进行比较。以ps位点为参照,上游
‑
3位倾向于精氨酸,
‑
1位同样也发现高比例的精氨酸和赖氨酸。上游
‑
2位上则偏向丙氨酸和苯丙氨酸。多肽的羧基端也表现出对氨基酸类型的轻微倾向性,主要体现为 1位的丙氨酸, 2位的苯丙氨酸有比较大的比例。这些不同位点上的氨基酸的偏好性与所使用的多肽文库数据高度一致。其次,评估了多肽文库数据与实验数据的总体相关性达到0.786,这表明建立的14
‑3‑
3与配体亲和力的定量构效关系模型得到了准确的预测。
[0090]
关于预测14
‑3‑
3蛋白与配体的亲和力,利用依据14
‑3‑
3多肽文库数据构建的定量构效关系模型预测所有可能序列组合的磷酸化三肽配体与14
‑3‑
3亚型的相对亲和力。预测得到的数据依据不同亚型进行数据标准化处理,使其平均值为0,方差为1,这样方便在不同亚型之间进行比较。标准化后的数据被视为14
‑3‑
3蛋白与配体亲和力的预测结果,并用于后续分析。标准化数据的数值范围在n库中是
‑
4.27到3.09,在c库中是
‑
4.46到3.67。收集了以往报道中经过生物实验所验证的与14
‑3‑
3蛋白有直接相互作用的哺乳动物来源的蛋白,并提取了14
‑3‑
3蛋白上与ps发生相互作用的周边位点,与本技术中的相应预测结果进行了比较,如图4。在酵母双杂交实验中发现人类细胞中的膜蛋白成分adam22与不同14
‑3‑
3亚型发生相互作用。在adam22蛋白序列中找到了两个磷酸化位点与前文中所提及的保守序列模式rsxpsxp一致。不过这两个磷酸化位点在 2位上都缺乏脯氨酸。之前有报道证明 2位上的脯氨酸替代为丙氨酸对14
‑3‑
3蛋白和配体之间的亲和力影响十分有限。所以可能 2位上的脯氨酸不关键。通过全部磷酸化配体的预测结果发现,14
‑3‑
3亚型之间具有高度保守的亲和力。这些被广泛研究的14
‑3‑
3蛋白结合配体中的磷酸化位点,例如c
‑
raf
‑
1和a
‑
raf中的rst、cdc25a中的rds和rps、pkc
‑
ε中的raa、pctaire
‑
2中的rak、mt中的rsh、th中的rha、tph中的rhs、a20中的rsk,能够匹配上保守序列模式rsxpsxp,在使用n库模型的预测结果中都显示较高的亲和力(标准化亲和力值大于1.5)。但是,cdc25c上的rsp位点是个例外。14
‑3‑
3蛋白通过与cdc25c发生相互作用参与细胞周期的调节过程。cds25c中已知磷酸化位点216上的多肽序列是rsppsmpe。虽然该序列与保守模式rsxpsxp相匹配,但是预测中发现氨基端rsp亲和力很低。有趣的是,羧基端mpe在预测中具有较高的亲和力。这暗示cdc25c与14
‑3‑
3蛋白的相互作用的机制可能与其他配体不同,并非依赖于氨基端而是羧基端。这也说明氨
基端和羧基端之间存在不平衡的配体亲和力。此外,能够匹配另外一种保守序列模式pxxpsxp的多肽序列,例如cdc25a中的rih和rfq、pkcγ中的cvr、irs
‑
1中的ptr、ck
‑
8中的syt、picalm中的lyr也显示出较高的配体亲和力预测结果。
[0091]
利用定量构效关系建模的方法获得14
‑3‑
3蛋白配体亲和力的预测模型,并获得全部配体亲和力的数据。将标准化的14
‑3‑
3配体亲和力数据,利用了二维层次聚类的方法进行了进一步的分析,如图5。从图中看到,聚类结果中没有发现在亲和力数据中明显的亚型特异性配体出现。但是有的亚型确实会显示出与其他亚型不同的亲和力模式。这可能与亚型之间在结构上的相似程度比较高有关。通过聚类结果确定了在氨基端配体中存在一组具有高结合亲和力的多肽序列,该组多肽的数量为253个。在图5中用框线圈出。该组多肽配体的亲和力值明显高于所有亚型的其他数据(p<0.0005)。通过序列分析发现该组多肽集中在保守模式rsxpsxp中。
[0092]
目前有多种类型的氨基酸描述符可以从已发表文献或数据库中获得。为了提高建模的性能,收集并测试了定量构效关系建模中常用的几类描述符z
‑
scales,vhse,hesh,g8和dpps。每一种类型的描述符都有其优势和局限性。其中z
‑
scales在预测肽活性的研究中广泛使用且非常有效,但也有报道在短肽序列建模中有局限性。虽然vhse和z
‑
scale是由不同种类的理化参数构建的,但它们都包含三类相互作用(疏水、静电和空间位阻)。以前的研究发现,使用vhse和z
‑
scale产生的结果不同。而在ace抑制剂、苦味肽、缓激肽的活性预测中,使用vhse描述符可以取得较好的结果。这表明vhse在描述蛋白质相互作用中的结构差异性上具有优势。与vhse(n=50)相比,dpps(n=171)和hesh(n=119)考虑了更多的物理化学参数。在dpps和hesh中额外考虑了氢键的相互作用。这两种类型的描述符已经被用于在不同的预测方法下建立一些的模型中。即使vhse和hesh是相对新发步的描述符,在初步的研究中,由vhse和hesh建立的模型质量(r2和rmse)也没有得到提高。在本技术的初步测试中,用dpps构建的模型性能优于其他描述符。因此,本技术中最终选择dpps作为氨基酸描述符。同时,也评估了各种较流行被采用的回归方法(包括svm、pls、mlr)的模型质量。这些方法建立的模型质量略有不同,但可以确认elastic net模型具有更好的拟合能力。因此,本技术中采用elastic net方法进行建模。在所使用的多肽文库数据中所使用十种氨基酸只占氨基酸全部种类的一半,尤其是在 /
‑
3位点只有五种。可以看出,基于多肽文库实验中存在的这种检测数量上局限性,很难得到关于14
‑3‑
3配体结合偏好性的全部数据。定量构效关系方法提供的计算方法帮助扩展对14
‑3‑
3蛋白相互作用的认识。根据模型中的参数,每个残基位点上重要的物理化学属性可以确定,氨基端残基的疏水特性对配体亲和力有重要贡献。14
‑3‑
3配体中保守的序列模式在的最终预测结果中得到了验证。确定了一些具有高亲和力的磷酸化配体序列,这些序列在未来的研究中具有很大的开发潜力。经过进一步的生物实验筛选和验证,这些以前未被发现的磷酸化配体有可能被进一步开发,作为干扰14
‑3‑
3相关途径的抑制剂或药剂用于一些癌症的靶向治疗上。
[0093]
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。