1.本发明属于生物医学领域,具体涉及一种构建骨髓增生异常综合征转白基因预测模型的方法。
背景技术:
2.骨髓增生异常综合征(myelodysplastic syndromes,mds)是一组起源于造血干细胞的临床常见血液系统恶性肿瘤,以难治性血细胞减少及无效造血以及高风险向急性髓细胞性白血病(acute myeloid leukemia,aml)进展为特征。患者有转化为白血病的可能。但由于个体异质性,不同患者白血病转化率及时间各异,故而临床上迫切需要有良好预测作用的指标。
3.与骨髓增生异常综合征白血病转化相关的因素主要包括性别、年龄等患者因素,以及疾病分型、血细胞数、基因突变、基因表达及表观遗传改变等疾病因素。目前临床常用的国际预后积分系统(ipss)、who分型预后积分系统(wpss)和修订的国际预后积分系统(r
‑
ipss)等系统从骨髓原始细胞比例、流式细胞学、细胞遗传学改变及血细胞减少程度等方面对患者进行预后评分并分组可对白血病转化进行预测。其中,细胞遗传学异常在分型分组中所占比重日益加重,如临床常见的del(5q)、del(20q)、
‑
7、 8、
‑
y等已被应用于预后分期。
4.然而值得注意的是在临床实践中,患者的临床表现及治疗效果差异仍较大,即便在同一分型和同一预后组中亦然。这表明目前纳入分型分级标准的临床和细胞遗传特征对于揭示mds的疾病本质尚且不足,新的分子层面的研究亟待开展以助于进一步深入了解疾病的致病机制,从而更精准预测mds的白血病转化率。
技术实现要素:
5.本发明的目的在于提供一种构建骨髓增生异常综合征转白基因预测模型的方法。
6.本发明的目的通过下述技术方案实现:
7.一种构建骨髓增生异常综合征转白基因预测模型的方法,包括以下步骤:
8.(1)收集高危mds患者的样本及其预后数据;提取样本dna并测序,获得突变谱;将高危mds患者的样本作为训练样本;
9.所述的高危mds患者是依据ipss分型确定的;
10.所述的样本为骨髓穿刺样品、血液,或其他组织;
11.所述测序的方法包括sanger测序、arms
‑
pcr(amplification refractory mutation system pcr)、mass
‑
pcr(mutation
‑
selected amplification specific system)、全基因组测序、全外显子测序以及小队列靶向测序;
12.所述的突变包括错义突变、无义突变、框移插入、框移缺失、非框移插入,非框移缺失及剪切位点突变,排除内含子变异及同义突变;
13.(2)利用oncodriveclust函数和dndscv函数,将步骤(1)获得的突变谱与现有高危mds患者dna测序数据库比对,将两种方法都比对到的基因定义为高危mds组驱动基因;
14.所述的高危mds组驱动基因为cbl、ezh2、runx1、idh1、asxl1、tet2、tp53、srsf2、idh2和jak2;
15.(3)将训练样本中的高危mds组驱动基因选取出,并且进行突变标记,当存在突变时标记为a1,不存在突变时标记为a2;以各训练样本的高危mds组驱动基因突变标记作为输入,对svm分类器模型进行训练,完成模型的构建;
16.所述步骤(3)中,a1为1,a2为0,或者a1为1,a2为0;
17.svm分类器模型可以选取0.5为阈值对样本进行预测,当样本预测值≥0.5,预测该样本为高风险转白;当样本预测值<0.5,预测该样本为低风险转白;
18.所述步骤(3)中,优选通过sample函数随机从入组高危mds组样本中选取70%患者作为训练集,将其驱动基因的突变标记作为输入,对svm分类器模型进行训练,再用剩余30%样本的预测值进行验证;
19.所述步骤(3)中,针对各训练样本,还提取出临床信息,包括原始细胞数、血细胞计数、细胞遗传学异常等指标,在训练时,将训练样本的高危mds组驱动基因突变与临床信息融合后作为输入,对svm分类器模型进行训练。
20.优选地,在步骤(2)获取高危mds组驱动基因后,利用lasso多重回归方法得到bonferroni校正后与白血病转化显著相关的突变基因,建立改进的wgrs模型:
21.wgrs=β1s1 β2s2 ,
…
, βnsn a;
22.a为常数;s1至sn为与白血病转化显著相关的各突变基因,β1至βn为对应权重;
23.根据改进的wgrs模型进行转白风险预测,预测得分<0.5为高风险转白,预测得分>0.5为低风险转白;
24.优选地,与白血病转化显著相关的突变基因为:cbl、ezh2、idh1和tp53;
25.所述改进的wgrs模型为:
26.wgrs=
‑
0.3459*cbl
‑
0.1989*ezh2
‑
0.2663*idh1 0.2174*tp53 0.5996。
27.本发明相对于现有技术具有如下的优点及效果:
28.相对于已有的针对骨髓增生异常综合征白血病转化的预测指标,本发明的模型构建方法从分子层面对疾病转白风险进行预测,不局限于骨髓增生异常综合征亚型的不同,对于所有成人患者可得到较精确的预测,对于高危患者进行早期干预,延缓疾病进展,且有助于后续治疗靶向药物的选择,有较高临床实用性。
附图说明
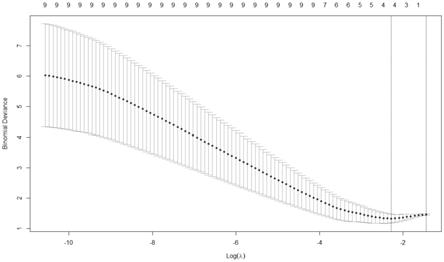
29.图1是实施例中lasso回归最小cp值的选择图,最小cp值为4。
30.图2是实施例中lasso回归中不同自变量选入顺序;不同的线代表不同自变量,竖线对应于lasso中迭代的次数,对应的系数值不为0的自变量即为选入的自变量。
31.图3是实施例中lasso回归模型的auc曲线图。
具体实施方式
32.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
33.实施例
34.骨髓增生异常综合征转白早期预测模型的建立
35.1)收集广东省人民医院血液科2018年1月至2019年12月的22例髓系肿瘤患者治疗前的骨髓穿刺样本,入组患者均根据ipss分型被诊断为高危mds患者,其中11例在随访期间进展为白血病,其余11例未进展为白血病。
36.2)收集20ng骨髓穿刺液提取基因组dna,应用ion proton
tm sequencer进行上机测序。对所得数据分别通过samtools
‑
1.8和picard
‑
2.19等软件进行排序、去除pcr重复和构建索引。接着,利用bcftools mpileup联合bcftools call进行变异检测(call variation)。
37.3)变异识别软件bcftools找到的变异为以(vcf,variant call format)格式储存,以qual<20and mq<40为指标过滤低质量值数据。并用refgene、cytoband、avsnp150、esp6500siv2_all、1000g2015aug_all、1000g2015aug_eas、dbnsfp30a、cosmic70、exac03、clinvar_20140929等数据库注释变异及氨基酸突变分析。
38.4)驱动基因:利用cbioportal数据库中mskcc数据集153例高危mds患者dna测序数据,通过r包“maftools”中oncodriveclust函数及r包“dndscv”进行驱动基因的检测(结果如表1和表2所示)。将两种方法检测到的共有驱动基因定义为高危mds组驱动基因,并应用于后续分析。
39.汇总表1和表2,高危mds组驱动基因为cbl、ezh2、runx1、idh1、asxl1、tet2、tp53、srsf2、idh2和jak2。
40.5)入组的22例高危mds患者驱动基因的突变情况如下(如表3所示);
41.表3
42.amlcblezh2runx1idh1asxl1tet2tp53srsf2idh2jak2y1000000000y1100000000y0100000000y0110100000y0000100000y0000010000y0100000000y1010100000y1110000000y0001000010y0010000100n0000010000n0000100001n0100000000n0000111000n0000011001n0010000100n0010001000
n0010000000n0010000000n0000000010n0000010000
43.注:y指该患者在随访期内发生白血病进展,n指该患者在随访期内未发生白血病进展;“1”指该样本在该基因发生突变,“0”表示该样本在该基因未发生突变
44.6)利用r 3.6.1中sample函数随机选取其中14例mds样本的10个基因突变有无作为训练集,利用r包“e1071”进行svm分类器模型训练,根据训练集结果选取0.5为预测阈值,将样本分为疾病转白和疾病未转白两组(如表4所示)。
45.表4
46.样本svm预测值预测结果临床结果一致性s10.0516901低风险进展未转白一致s20.052018222低风险进展未转白一致s30.180939376低风险进展未转白一致s40.19531375低风险进展未转白一致s50.344135252低风险进展未转白一致s60.750653047高风险进展未转白不一致s70.750653047高风险进展转白一致s80.947917056高风险进展未转白不一致s90.947917056高风险进展转白一致s100.947917056高风险进展转白一致s110.948101223高风险进展转白一致s120.948182257高风险进展转白一致s130.948243325高风险进展转白一致s140.948380951高风险进展转白一致
47.7)在剩余8例样本中验证此模型的分类效能(如表5所示)。
48.表5
49.样本svm预测值预测结果临床结果一致性s10.36484287低风险进展未转白一致s20.441575904低风险进展未转白一致s30.461959559低风险进展未转白一致s40.461959559低风险进展未转白一致s50.461959559低风险进展转白不一致s60.624852621高风险进展转白一致s70.654242169高风险进展转白一致s80.948182257高风险进展转白一致
50.8)为了进一步确认mds中与白血病转化相关性更强的基因,利用lasso多重回归方法得到bonferroni校正后与白血病转化显著相关的突变基因,为:cbl、ezh2、idh1和tp53,建立改进的wgrs模型(如图1
‑
图3所示);把每个纳入的突变基因都当作变量s,根据得到的
每个基因的权重值β,于是改进的wgrs模型表示为各变量与自己的权重乘积之和;
51.wgrs=
‑
0.3459*cbl
‑
0.1989*ezh2
‑
0.2663*idh1 0.2174*tp53 0.5996。
52.上述步骤7)的实现代码可以如下:
53.library(lars)
54.svm51$aml=as.numeric(svm51$aml)
55.x=as.matrix(svm51[,2:10])
[0056]
y=as.matrix(svm51[,1])
[0057]
lar1<
‑
lars(x,y,type="lasso")
[0058]
plot(lar1)
[0059]
summary(lar1)
[0060]
lar1$cp[which.min(lar1$cp)]
[0061]
lar1$beta
[0062]
coef<
‑
coef.lars(lar1,mode="step",s=5)
[0063]
coef[coef!=0]
[0064]
predict(lar1,data.frame(cbl=0,ezh2=0,tp53=0,idh1=0,asxl1=0,srsf2=0,idh2=0,runx1=0,tet2=0),s=5)
[0065]
根据所改进模型进行转白风险预测,预测得分<0.5为高风险转白,预测得分≥0.5为低风险转白,如表6所示。
[0066]
表6
[0067]
amlcblezh2idh1tp53得分预测值一致性y10000.2537y一致y11000.0548y一致y01000.4007y一致y01000.4007y一致y01000.4007y一致y10000.2537y一致y11000.0548y一致y00100.3333y一致n01000.4007y不一致n00010.817n一致n00010.817n一致n00010.817n一致
[0068]
注:y指该患者在随访期内发生白血病进展,n指该患者在随访期内未发生白血病进展;“1”指该样本在该基因发生突变,“0”表示该样本在该基因未发生突变
[0069]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
[0070]
[0071]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。