1.本发明涉及薄层鉴别领域,具体涉及一种薤白供试品的薄层鉴别方法。
背景技术:
2.薤白是百合科植物小根蒜allium macrostemon bge.或薤allium chinense g.don的干燥鳞茎。薤白是我国传统常用中药材,性温,味辛、苦。归心、肺、胃、大肠经。具有抗肿瘤、抗氧化、抑制血小板凝聚、降脂、抑菌和解痉平喘等功效,可用于治疗冠心病和心绞痛、支气管哮喘、高血脂以及心肌缺血等疾病。薤白药材具有毒性低、安全范围广、药源丰富、价格低廉的优点,故加强对薤白药用制剂的研究及开发具有良好的前景。
3.薤白化学成分复杂,有甾体皂苷、挥发油、含氮化合物、酸性成分、多糖、无机元素等多种成分。其中甾体皂苷是薤白的主要活性成分之一,具有抑制凝血、抗肿瘤、平喘等多种药理作用,也是薤白药效的物质基础。由于薤白水提物、配方颗粒是薤白药材经一系列工艺制成,其中一些工艺会破坏原药材的性状特征,为鉴别已被破坏性状的中药水提物、配方颗粒,需要一种具有很好的区分和鉴定能力且操作更加简便、快捷的鉴别方法,以便能够更好地控制质量。
4.而薄层色谱鉴别方法具有操作简便、快捷的优势,是一种较好的鉴别方法。对于薤白而言,目前药典记载了该薤白药材的薄层鉴别方法,具体的,以正己烷为溶剂、正己烷
‑
乙酸乙脂(10:1)为展开剂,采用硅胶板g薄层板,喷以10%硫酸乙醇溶液,在105℃加热显色,置紫外光灯(365nm)下检视。
5.但在上述药典中记载的薤白药材的薄层鉴别方法中,其检视得到的薄层具有显示的色斑模糊,分离效果不好的问题。同时,检视得到的色谱信息少,尤其是对于薤白水提物、配方颗粒而言,信息更少。并且,现有薄层色谱鉴别方法中,在展开后需要采用显色剂进行处理,导致对操作人员及环境并不友好。
技术实现要素:
6.因此,本发明要解决的技术问题在于,克服现有技术中的薄层色谱鉴别存在显示的色斑模糊、分离效果不好、色谱信息少的缺陷;从而提供具有更多色谱信息、分离效果佳、色斑清晰且对操作人员更加友好的一种薤白供试品的薄层鉴别方法。
7.一种薤白供试品的薄层鉴别方法,包括:
8.取薤白药材、水提物或配方颗粒,制备成供试品溶液;
9.将供试品溶液点于硅胶g薄层板上,以体积比为(5~6):(3.5~4):(1~1.2):(0.8~1)的石油醚
‑
乙酸乙酯
‑
二氯甲烷
‑
甲酸为展开剂,展开,取出,晾干,紫外灯检视。
10.所述石油醚为沸点温度为60
‑
90℃的中沸点石油醚。
11.所述供试品溶液的制备过程为:
12.取薤白药材、水提物或配方颗粒,加入水和盐酸,提取后,放冷,用乙酸乙酯振摇提取,浓缩后即得供试品溶液。
13.所述盐酸与水的体积比为3:(30
‑
50)。
14.所述乙酸乙酯的加入量为水体积的1
‑
1.5倍;
15.浓缩后的体积为原体积的2
‑
10%。
16.所述提取的方式为加热回流;提取时间为30
‑
90min。
17.还包括对照药材溶液的鉴别,对照药材溶液的制备方法与供试品溶液的制备方法相同。
18.所述紫外灯检视的波长为365nm。
19.所述薤白供试品为薤白药材、薤白饮片、薤白水提物或薤白配方颗粒。
20.所述薤白水提物为采用水为提取溶剂进行提取获得提取液、浓缩液或干燥粉。
21.本发明技术方案,具有如下优点:
22.1.本发明提供的了一种薤白供试品的薄层鉴别方法,本方法以体积比为(5~6):(3.5~4):(1~1.2):(0.8~1)的石油醚
‑
乙酸乙酯
‑
二氯甲烷
‑
甲酸为展开剂,展开并检视后,能够获得相比药典中更多的薄层色谱信息斑点,薄层信息丰富;并且,显示出的斑点清晰且分离度好,重性现好。
23.2.本发明提供的薄层鉴别方法,依据薤白中甾体皂苷类薄层鉴别方法制定,可以更好的保留薤白中水溶性成分的多信息,进而保证薤白水提物或薤白配方颗粒中具有更多的薄层信息,保证了薤白药材、水提物及配方颗粒工艺工程中的质量可控性,保证临床用药疗效;同时,也为薤白以水提取入药的多种制剂提供了薄层鉴别参考图和方法。
24.3、本发明方法不用显色剂,直接置紫外光(365nm)下检视,即可显示出清晰的斑点。与现有鉴别方法相比,该检视过程简便、快捷,减少了对操作人员身体健康的损害、及环境的污染。
附图说明
25.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
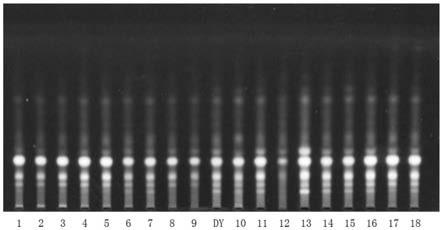
26.图1是本发明实施例1中18批薤白水提物的薄层鉴别检视结果图;
27.图2是本发明实施例1中三批薤白药材的薄层鉴别检视结果图;
28.图3是本发明实施例2中供试品溶液在不同点样量下的薄层鉴别检视结果图;
29.图4是本发明实施例3中供试品溶液在常温低湿条件下的薄层鉴别检视结果图;
30.图5是本发明实施例3中供试品溶液在低温常湿条件下的薄层鉴别检视结果图;
31.图6是本发明实施例3中供试品溶液在高温高湿条件下的薄层鉴别检视结果图;
32.图7是本发明实施例3中采用merck kgaa的硅胶g板的薄层鉴别检视结果图;
33.图8是本发明实施例3中采用烟台市芝罘黄务硅胶开发试验厂的硅胶g板的薄层鉴别检视结果图;
34.图9是本发明实施例3中采用烟台市化学工业研究所的硅胶g板的薄层鉴别检视结果图;
35.图10是本发明实施例4中采用的3批供试品溶液的薄层鉴别检视结果图;
36.图11是本发明实施例5的薄层鉴别检视结果图;
37.图12是本发明对比例1的薄层鉴别检视结果图;
38.图13是本发明对比例2的薄层鉴别检视结果图。
具体实施方式
39.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
40.仪器:camag薄层成像系统,me104e电子天平(梅特勒
·
托利多),jy20002电子天平(梅特勒
·
托利多),bsa124s电子天平(赛多利斯科技仪器(北京)有限公司),bt25s电子天平(赛多利斯科技仪器(北京)有限公司),dzkw
‑
4恒温水浴锅(北京中兴伟业仪器有限公司),kq
‑
500db超声波清洗器(昆山市超声仪器有限公司),硅胶g板(merck kgaa,烟台市化学工业研究所,烟台市芝罘黄务硅胶开发试验厂)。
41.试药:薤白对照药材(批号:121130
‑
201906,中国食品药品检定研究院);
42.薤白水提物:分别取批号为200618
‑
725800
‑
01、200529
‑
724400
‑
02、200610
‑
713800
‑
03、200703
‑
725800
‑
04、200703
‑
725800
‑
05、200703
‑
043100
‑
06、200703
‑
043100
‑
07、200703
‑
725800
‑
08、200703
‑
043100
‑
09200703
‑
725800
‑
10、200703
‑
043100
‑
11、200703
‑
727000
‑
12、200703
‑
710000
‑
13、200703
‑
710038
‑
14、200703
‑
712000
‑
15、200703
‑
710000
‑
16、200703
‑
714100
‑
17、200703
‑
724400
‑
18的薤白饮片,置于砂锅中,浸泡30分钟,一煎加入饮片量6倍水,武火煮沸后,文火煎煮30分钟,趁热过滤,迅速冷却,备用;二煎加饮片量4倍水,武火煮沸后,文火煎煮20分钟,趁热过滤,迅速冷却备用;合并滤液,浓缩(65℃),浓缩至料液比约为1:1(相对密度为1.05
‑
1.10),冷冻干燥,即得对应批号的水提物。
43.薤白药材:yc200618
‑
725800
‑
01、yc200529
‑
724400
‑
02,yc200610
‑
713800
‑
03。
44.试剂:石油醚(60~90℃)、乙酸乙酯、二氯甲烷、盐酸、甲酸均为分析纯,水为蒸馏水。
45.实施例1
46.一种薤白供试品的薄层鉴别方法,具体过程如下:
47.1、供试品溶液的制备:
48.供试品溶液的制备:取薤白水提物0.5g,研细,加水30ml,盐酸3ml,加热回流1小时,放冷,用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,浓缩至5ml,作为供试品溶液。
49.供试品溶液的制备:取薤白药材1.5g,研细,加水50ml,盐酸3ml,加热回流1小时,放冷,用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,浓缩至5ml,作为供试品溶液。
50.对照药材溶液的制备:取薤白对照药材1.5g,研细,加水50ml,盐酸3ml,加热回流1小时,放冷,用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,浓缩至5ml,作为对照药材溶液。
51.2、薄层鉴别的过程:
52.吸取供试品溶液2μl,对照药材1μl,分别点于同一硅胶g薄层板上,以5:3.5:1:0.8的石油醚(60~90℃)
‑
乙酸乙酯
‑
二氯甲烷
‑
甲酸为展开剂,展开,取出,晾干,置365nm紫外
光灯下检视。
53.本实施例中对上述18批的薤白水提物以及3批次的薤白药材制备得到的供试品溶液以及对照药材溶液分别进行检视,检视结果如图1和图2所示。图1中,编号1
‑
18依次对应上述18批薤白水提物,dy对应薤白对照药材;图2中,编号1
‑
3依次对应上述3批薤白药材,dy对应薤白对照药材。
54.实施例2
55.本实施例与实施例1的区别在于,本发明采用的供试品溶液为批号200618
‑
725800
‑
01、200529
‑
724400
‑
02、200610
‑
713800
‑
03的薤白水提物采用实施例1中相同的方式制备得到,且供试品溶液和对照药材溶液的点样量不同,具体设置为:点样量分别为1μl、2μl、3μl,结果见图3所示。
56.该图3中,编号1
‑
3依次对应1μl、2μl、3μl的薤白水提物200618
‑
725800
‑
01,编号4
‑
6依次为1μl、2μl、3μl的薤白水提物200618
‑
725800
‑
02,编号7
‑
9依次对应1μl、2μl、3μl的薤白水提物200618
‑
725800
‑
03,编号10
‑
12依次对应1μl、2μl、3μl的薤白对照药材。
57.通过对点样量的考察,薤白水提物、薤白对照药材点样量分别为2μl、1μl时,薄层色谱斑点清晰,分离度佳。故供试品溶液、对照药材溶液最佳点样量为2μl、1μl。
58.实施例3
59.本实施例对实施例1中的薄层鉴别进行耐用性验证,具体包括:
60.1、不同温湿度的考察
61.采用实施例1中制备得到的供试品溶液(薤白水提物:200618
‑
725800
‑
01、200529
‑
724400
‑
02、200610
‑
713800
‑
03)、对照药材溶液,分别在常温低湿(t:29℃,rh:10%)、低温常湿(t:4℃,rh:60%)、高温高湿(t:29℃,rh:80%)条件下,采用实施例1相同的方法进行展开,取出,晾干,置紫外光灯(365nm)下检视,结果见图4
‑
6。该图4
‑
6中编号1
‑
3分别对应薤白水提物200618
‑
725800
‑
01、薤白水提物200529
‑
724400
‑
02、薤白水提物200610
‑
713800
‑
03,编号4对应薤白对照药材。
62.2、不同薄层板的考察
63.采用实施例1中制备得到的供试品溶液(薤白水提物:200618
‑
725800
‑
01、200529
‑
724400
‑
02、200610
‑
713800
‑
03)、对照药材溶液,分别采用merck kgaa、烟台市芝罘黄务硅胶开发试验厂、烟台市化学工业研究所三种厂家的硅胶g板,按照实施例1中相同的条件进行展开,取出,晾干,置紫外光灯(365nm)下检视,结果见图7
‑
9。该图7
‑
9中编号1
‑
3分别对应薤白水提物200618
‑
725800
‑
01、薤白水提物200529
‑
724400
‑
02、薤白水提物200610
‑
713800
‑
03,编号4对应薤白对照药材。
64.实施例4
65.本实施例与实施例1的区别在于,本发明采用实施例1相同的条件进行3批薤白配方颗粒的检测;该薤白配方颗粒的制备方法为:取薤白饮片,加水煎煮,滤过,滤液浓缩成清膏,干燥(或干燥,粉碎),加辅料适量,混匀,制粒,即得。采用该薤白配方颗粒制备成供试品溶液的过程与实施例1中薤白水提物的制备方法完全相同;其他条件也与实施例1完全相同,3批薤白配方颗粒的检测结果如图10所示。
66.该图10中编号1
‑
3顺次对应薤白配方颗粒kl200618
‑
725800
‑
01、薤白配方颗粒kl200529
‑
724400
‑
02、薤白配方颗粒kl200610
‑
713800
‑
03,编号4对应薤白对照药材。
67.实施例5
68.本实施例与实施例4的区别在于,本实施例中展开剂的比例、供试品及其点样量不同,其他均与实施例4相同。具体设置为:展开剂采用体积比为6:4:1.2:1的石油醚
‑
乙酸乙酯
‑
二氯甲烷
‑
甲酸,采用薤白配方颗粒kl200618
‑
725800
‑
01作为供试品,供试品溶液的点样量分别为5μl和10μl,检测结果如图11所示。
69.该图11中编号1
‑
2顺次对应5μl和10μl的薤白配方颗粒kl200618
‑
725800
‑
01,编号3对应薤白对照药材。
70.对比例1
71.本实施例采用《中国药典》2015年版一部的方法对薤白水提物以及薤白对照药材进行薄层色谱检测,具体过程如下:
72.供试品溶液的制备:取薤白水提物4g,加正己烷20ml,超声处理20分钟,滤过,滤液挥干,残渣加正己烷1ml使溶解,作为供试品溶液。
73.对照药材溶液的制备:取薤白对照药材4g,加正己烷20ml,超声处理20分钟,滤过,滤液挥干,残渣加正己烷1ml使溶解,作为对照药材溶液。
74.薄层色谱条件:吸取供试品与对照药材两种溶液各10μl,分别点于同一硅胶g薄层板上,以正己烷
‑
乙酸乙脂(10:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外(365nm)下检视,结果如图12所示。图12中,编号1对应薤白水提物200618
‑
725800
‑
01;编号2对应薤白对照药材。
75.对比例2
76.本实施例与对比例1的区别在于,薄层色谱检测的过程不同,具体过程如下:
77.供试品溶液的制备:取薤白水提物4g,加正己烷20ml,超声处理20分钟,滤过,滤液挥干,残渣加正己烷1ml使溶解,作为供试品溶液。
78.对照药材溶液的制备:取薤白对照药材4g,加正己烷20ml,超声处理20分钟,滤过,滤液挥干,残渣加正己烷1ml使溶解,作为对照药材溶液。
79.薄层色谱条件:吸取供试品与对照药材两种溶液各10μl,分别点于同一硅胶h薄层板上,以正己烷
‑
乙酸乙脂(10:1)为展开剂,展开,取出,晾干,用碘蒸气熏至斑点显色清晰,日光下检视,结果如图13所示。图13中,编号1对应薤白水提物200618
‑
725800
‑
01;编号2对应薤白对照药材。
80.显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。