1.本发明属于生物医药技术领域,具体涉及一种黄体酮缓释组合物及其应用。
背景技术:
2.黄体酮是由卵巢分泌的一种天然孕激素,是目前临床黄体支持的首选药物,对女性子宫内膜有显著的保护作用。临床上市剂型主要有口服胶囊、凝胶栓剂、肌注油溶制剂等,其中,黄体酮口服制剂首过效应严重,口服生物利用度仅为10%,导致疗效不佳、副作用较大。因此,临床上普遍使用油溶液肌注。现有的黄体酮注射剂需每天注射,且为长期注射,每天注射黄体酮极为不便,同时油溶液注射时极易引起患者不适,如疼痛、局部红肿、过敏等症状。而研发一种释放周期为几天或几周的黄体酮长效制剂可以使患者在一个临床使用疗程只需注射一次,显著减少给药次数,克服油注射剂长期高频次注射会导致注射部位产生硬结等其他症状而不利于注射部位组织健康的缺点,从而改善了病人用药顺应性,更适应临床需求。
3.注射型原位凝胶(in situ gel)指在溶液状态下给药,在生理环境液体原位转化为固体或半固体储库的一类制剂。原位胶凝组合物有几个优点:它们可用简易的制备方法生产并以方便注射的液体形式储存、延长释药周期、无需外科手术植入和取出即可在体内形成植入物。其中沉淀型原位凝胶由于其良好的成形和缓释效果在注射型制剂应用最为广泛,其基质材料主要为聚乳酸(pla)、丙交酯乙交酯共聚物(plga)、聚己内酯(pcl),因为它们已获得美国食品和药物管理局(fda)的批准并且长期应用于临床。其中plga由于其具有无定形的结构、已知的降解速率和良好的生物相容性作为该制剂的缓释基质具有独特的优势,目前已有多个以plga作为基质的原位凝胶产品上市。
4.现有的plga体系的应用也有下列限制。第一、释放速度慢:在药物缓控释领域,对不同的药物要求其载体材料具有不同的释药速度,而由于plga表面疏水性强,形成的固体凝胶结构致密,药物(特别是疏水性药物)包裹在其中通过扩散、溶出或聚合物溶蚀等途径释放缓慢。已上市产品的药物释放周期一般为几个月(美国专利6,565,874、6,528,080、6,461,631,6,395,293、4,938,763、5,077,049、和20130210853),例如(释放时间为1、3、4、6个月)、(释放时间为1个月)、(释放时间为1个月)等。第二、降解时间长:不同型号plga降解时间在1~6个月,仅依靠plga均聚物的分子量和分子量分布调节降解速度具有很大的局限性,单独的均聚物原位凝胶不能满足释放周期小于一个月的药物的释放。第三、载药量限制:由于安全性高、水相相容性高,目前上市的plga体系溶剂均为n
‑
甲基
‑2‑
吡咯烷酮(nmp),但药物在溶剂中的溶解度和原位凝胶注射体积的限制(一般小于2ml),plga体系主要用于小剂量药物或在nmp中溶解度高的药物,例如(每月给药7.5mg)、(每月给药90mg或120mg)、(丁丙诺啡在nmp的溶解度大于300mg/ml)。而对于黄体酮这种剂量大(每月给药300mg~600mg)、在两亲性溶剂溶解度低的药物,现有的plga体系由于药物释放周期过长,无法适应于黄体酮的临床需求;同时,该制
剂体系还存在突释现象较为严重的缺点,严重影响其长效制剂释药平稳性,不利于产品质量控制。
技术实现要素:
5.本发明的目的是提供一种黄体酮缓释组合物及其应用。
6.为了实现上述发明目的,本发明采用以下技术方案:
7.一种黄体酮缓释组合物,原料包括:原料以质量百分数计包括:黄体酮1~30%,脂肪酸1~40%,溶剂40~80%,各组分的质量分数之和为100%;
8.所述脂肪酸选自硬脂酸、花生酸、软脂酸、月桂酸、肉豆蔻酸、山嵛酸、肉豆蔻酸、棕榈油酸、单油酸甘油酯、二油酸甘油酯、花生四烯酸或油酸;
9.所述溶剂选自n
‑
甲基吡咯烷酮(nmp)、甘油三醋酸酯、乳酸乙酯、甘油缩甲醛、苯甲酸苄酯、乙酸乙酯、peg 400、peg 600、苯甲醇、二甲亚砜、丙二醇、丙酮、乙醇、2
‑
吡咯烷酮或碳酸丙烯酯中的一种或几种。
10.优选地,所述脂肪酸选自硬脂酸、花生酸、软脂酸、月桂酸、肉豆蔻酸或山嵛酸。
11.优选地,所述溶剂为n
‑
甲基吡咯烷酮(nmp)、peg 400、peg 600、乳酸乙酯、三醋酸甘油酯、丙二醇或苯甲酸苄酯。
12.进一步地,所述组合物的原料还包括可生物降解聚合物,其用量以质量百分数计为1~40%。
13.所述可生物降解聚合物选自聚乳酸(pla)、丙交酯乙交酯共聚物(plga)、聚己内酯(pcl)、聚原酸酯(poe)、聚乳酸
‑
聚乙二醇嵌段共聚物(peg
‑
pla)、聚乙二醇
‑
聚乙交酯丙交酯嵌段共聚物(peg
‑
plga)、聚n
‑
异丙基丙烯酰胺或乙酸异丁酸蔗糖酯(saib)中的一种或几种。
14.优选地,所述可生物降解聚合物为丙交酯乙交酯共聚物,丙交酯乙交酯共聚物中丙交酯和乙交酯的摩尔比为(50~85):(15~50),所述丙交酯乙交酯共聚物的分子量为3000~40000道尔顿。
15.上述黄体酮缓释组合物在制备黄体酮注射剂中的应用。
16.本发明使用脂肪酸作为原位凝胶基质,注射到皮下或肌肉后,脂肪酸可以迅速包载药物形成固体植入物,而黄体酮可以与脂肪酸形成多孔网状交联结构,该黄体酮
‑
脂肪酸结合物在水性环境下缓慢溶出,其中的脂肪酸可以促进脂溶性药物的吸收,与plga凝胶相比,脂肪酸形成的凝胶释药速率大幅提高,同时可以采用任意比例的脂肪酸/plga组合,实现灵活调节药物释放速度。其次,plga表面疏水性强,形成的固体凝胶结构致密,而脂肪酸凝胶结构更加疏松,表面侵蚀更快,同时脂肪酸在体内可以通过β
‑
氧化等途径被代谢和吸收,相比plga代谢分解更快,制剂消除速率更快。该组合物克服现有plga原位凝胶由于释放周期长、降解速度慢、载药量限制无法适应释放周期小于1个月的药物的释放。
17.本发明可以通过调整脂肪酸的种类和比例,灵活调节药物释放速度,实现3~15天的释药周期;通过脂肪酸促进药物吸收,药物释放更加完全,从而适应于释药周期为3~15天、每日剂量大的药物的临床给药需求。本发明的组合物有更快的分解速度,制剂清除更快;具有更好的注射成形性,且可以显著改善原位凝胶制剂在注射初期的突释现象,有利于提高药物疗效。
附图说明
18.图1为实施例1中处方1(左)和对照例1(右)的体外成形外观。
19.图2为实施例3中1小时内原位凝胶的体外释放突释结果。
20.图3为实验例1中脂肪酸原位凝胶与plga原位凝胶的体外释放对比曲线。
21.图4为实验例3中脂肪酸原位凝胶(左)与plga原位凝胶(右)的体外降解速率对比结果。
22.图5为实验例5中脂肪酸原位凝胶的dsc结果。
23.图6为实验例6中脂肪酸原位凝胶的体内成形性结果。
具体实施方式
24.以下实施例是对本发明的进一步说明,但绝不是本发明范围的限制,下面参考实施例进一步详细阐述本发明,但是本领域技术人员应当理解,本发明并不限于这些实施例以及使用的制备方法。而且,本领域技术人员根据本发明的描述可以对本发明进行等同替换、组合、改良或修饰,但这些都将包括在本发明的范围内。
25.对照例1
26.取黄体酮100mg,溶于700mg n
‑
甲基吡咯烷酮(nmp)中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入plga(evonik corporation,rg502h)200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明溶液。
27.对照例2
28.取黄体酮100mg,溶于700mg的nmp,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入pla 200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
29.实施例1
30.脂肪酸缓释组合物的凝胶相变过程
31.按表1处方分别称取不同种类的脂肪酸200mg、黄体酮100mg、nmp 700mg,以及可生物降解聚合物50mg,加热搅拌2小时,得到澄清透明溶液。
32.将得到的溶液注入5ml pbs(ph=7.4)中,考察凝胶的体外成形性。结果如表1所示。
33.表1不同基质脂肪的制备及成形性考察
34.处方脂肪酸种类混合基质胶凝时间处方1硬脂酸无20s处方2花生酸无22s处方3软脂酸无23s处方4月桂酸无21s处方5肉豆蔻酸无22s处方6山嵛酸无25s处方7棕榈油酸无未能胶凝处方8硬脂酸plga35s处方9硬脂酸pla45s
处方10硬脂酸poe<60s处方11硬脂酸saib<60s对照例1无plga<60s
35.对照例1pbs缓冲液中有混浊液产生,说明凝胶在成形后有固体泄漏。而脂肪酸组在5
‑
30秒内即可成形完整,成形后无混浊液产生,处方1的体外成形外观如图1所示。
36.实施例2
37.不同种类溶剂的处方筛选
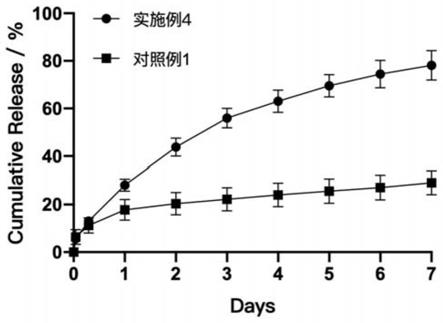
38.分别称取脂肪酸20%(w/w)、黄体酮10%(w/w),按表2分别称取不同种类的溶剂,加热搅拌3小时,得到澄清透明溶液。
39.将得到的溶液注入5ml pbs(ph=7.4),考察凝胶的体外成形性。结果如表2所示。
40.表2不同溶剂的处方筛选及成形性考察
41.处方溶剂溶液外观成形性处方170%nmp澄清透明成形完整处方270%peg
‑
400澄清透明成形完整处方370%peg
‑
600澄清透明成形完整处方470%乳酸乙酯澄清透明成形完整处方570%三醋酸甘油酯澄清透明成形完整处方630%丙二醇和40%nmp澄清透明成形完整处方750%苯甲酸苄酯和10%nmp澄清透明成形完整
42.由表2可知,所选溶剂均能制得成形完整的产品。
43.实施例3
44.不同浓度脂肪酸的抑制突释特性考察
45.将含10%(w/w)的黄体酮和不同浓度脂肪酸和plga混合基质的缓释组合物注射含0.5%sds
‑
10ml pbs缓冲溶液中(ph=7.4),在恒温振荡器(37℃,100rmp)中进行体外释放试验。在设定时间点取出10ml的缓冲液,并加入等体积的新鲜pbs缓冲液,计算黄体酮的累计释放率。结果如表3所示。
46.表3不同浓度脂肪酸的缓释特性考察
47.处方脂肪酸浓度(w/w)plga浓度(w/w)1小时突释对照例10%20%7.79%处方11%19%5.01%处方22%18%3.55%处方35%15%2.74%处方48%12%1.95%处方510%10%1.87%
48.如表3和图2可知,脂肪酸
‑
plga凝胶组具有更快的释放速率和抑制突释的特性。
49.实施例4
50.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入硬脂酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
51.实施例5
52.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入花生酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
53.实施例6
54.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入软脂酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
55.实施例7
56.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入月桂酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
57.实施例8
58.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入肉豆蔻酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
59.实施例9
60.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入山嵛酸200mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
61.实施例10
62.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入plga150mg和硬脂酸50mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
63.实施例11
64.取黄体酮100mg,溶于700mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入软脂酸150mg和peg
‑
plga50mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
65.实施例12
66.取黄体酮100mg,溶于600mg的nmp和100mg peg 600中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入花生酸150mg和pla 50mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
67.实施例13
68.取黄体酮100mg,溶于600mg的nmp和100mg peg 600中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入硬脂酸150mg和聚原酸酯50mg,无菌条件下磁力搅拌约1小时,至完全溶解,得到澄清透明液体,分装,密封,即得。
69.实施例14
70.取黄体酮100mg,溶于500mg的peg400和200mg的nmp中,经0.22μm微孔滤膜过滤得到药物溶液,然后在无菌条件下,加入硬脂酸200mg,无菌条件下磁力搅拌约1小时,至完全
溶解,得到澄清透明液体,分装,密封,即得。
71.实验例1
72.体外释放实验
73.将实施例4和对照例1的制剂注射在20ml西林瓶中,然后放置在10mlpbs缓冲溶液中(ph=7.4),在恒温振荡器(37℃,100rmp)中进行体外释放试验。在设定时间点取出10ml的缓冲液,并加入等体积的新鲜pbs缓冲液,计算黄体酮的累计释放率。
74.结果见图3,脂肪酸缓释组合物相比plga缓释组合物具有更快的释放速率和抑制突释的特性。
75.实验例2
76.体外释放实验
77.体外释放实验方法同实验例1,取对照例1、对照例2、实施例5、实施例6、实施例10、实施例14的制剂进行体外实验,结果如表4。
78.表4体外释放实验
79.处方7天累积释放百分率对照例132.12%对照例229.90%实施例575.81%实施例677.64%实施例1067.95%实施例1474.83%
80.由表4可知,脂肪酸缓释组合物相比plga缓释组合物具有更快的释放速率和抑制突释的特性。
81.实验例3
82.分解速率考察
83.取对照例1、实施例4、实施例5处方进行,然后放置在10ml pbs缓冲溶液中(ph=7.4),在恒温振荡器(37℃,100rmp)中进行体外降解试验。经过一定时间后取出制剂,干燥,称取剩余含量,计算分解速率。结果如表5和图4所示,脂肪酸缓释组合物相比plga缓释组合物具有更快的分解速率。
84.表5体外分解速率考察
85.处方分解速率(14天)对照例110%实施例463%实施例544%
86.实验例4
87.不同释放度考察
88.将制剂注射在20ml西林瓶中,然后放置在10mlpbs缓冲溶液中(ph=7.4),在恒温振荡器(37℃,100rmp)中进行体外释放试验,纪录固化胶凝时间点,在设定时间点取出10ml的缓冲液,并加入等体积的新鲜pbs缓冲液,计算黄体酮的累计释放率。结果见表6。
89.表6体外释放度考察
90.处方胶凝时间释放度(至80%时间)对照例1>60秒>30天实施例420秒7天实施例830秒5天实施例933秒6天实施例1035秒10天
91.实验例5
92.dsc考察
93.将实施例4的脂肪酸缓释组合物进行dcs考察,实施例4的dsc分析结果如图5所示,黄体酮原料药在131℃处有明显吸热峰,脂肪酸在72℃处有明显吸热峰,黄体酮和脂肪酸的物理混合物则分别出现黄体酮、脂肪酸的特征吸热峰,而黄体酮
‑
脂肪酸缓释组合物形成的原位凝胶在131℃处未见黄体酮特征吸热峰,且72℃处脂肪酸的特征吸热峰变为68℃和79℃双峰有变宽趋势,结果说明,黄体酮
‑
脂肪酸缓释组合物形成的原位凝胶中黄体酮与脂肪酸不是简单的物理混合,而是存在分子间作用力。
94.实验例6
95.体内成形性实验
96.取0.5ml实施例4的制剂,进行大鼠背部皮下注射,考察体内成形性,结果见图6。黄体酮
‑
脂肪酸缓释组合物可在体内成形,且成形性完整。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。