1.本发明涉及一种左酮咯酸的药物组合物及其制备方法,属于药物制剂技术领域。
背景技术:
2.酮咯酸氨丁三醇(ketorolac tromethamine)是一种非甾体类解热镇痛药,结构为较特殊的吡咯酸衍生物,最早由美国syntex公司开发上市。其通过抑制前列腺素(pg)的合成而达到止痛、抗炎和退热的作用,并非作用于阿片受体或激发体内阿片肽的释放。酮咯酸氨丁三醇可以缓解各种肌肉、软组织和关节的中毒疼痛,适用于需要阿片镇痛药的急性较严重疼痛的短期治疗,通常用于手术后镇痛,而不适用于轻度或慢性疼痛的治疗,该药无成瘾性。在标准的镇痛活性动物模型中,酮咯酸氨丁三醇的镇痛活性是阿司匹林的800倍,比消炎痛和萘普生强的多,优于保泰松。与阿片类镇痛剂相比,酮咯酸具有起效快、无成瘾性、无中枢神经系统损害、无呼吸抑制作用或便秘等不良反应、作用时间长的优点。酮咯酸与吗啡合用可降低吗啡用量,减少吗啡带来的不良反应和成瘾性。
3.目前国内临床上所用的酮咯酸氨丁三醇剂型主要有:片剂、胶囊剂,注射剂和滴眼液。美国还有鼻腔喷雾剂上市。此外酮咯酸的外用制剂如凝胶剂也逐渐获得研究人员的关注。
4.酮咯酸氨丁三醇注射液以其快速镇痛疗效好的特点,在临床术后镇痛等领域得到了广泛使用。目前国内外上市的酮咯酸氨丁三醇注射液主要有两种配方:一种为磷酸二氢钾为ph调节剂,以永信药品工业(昆山)有限公司的产品为代表;另一种则是大多数公司采用的、以乙醇作为稳定剂。但是,酮咯酸氨丁三醇注射液在传统的处方中含有乙醇作为助溶剂和稳定剂。在长期放置中,随着乙醇不可避免的挥发性,仍然有白点或结晶析出,影响可见异物及不溶性微粒等质量指标,在临床使用中存在安全隐患。另外,与头孢类抗生素合用时,可发生双硫仑反应,导致患者血压下降、心跳加速或心电图部分改变。因此,含有乙醇的酮咯酸氨丁三醇注射液具有安全隐患。
5.中国专利cn101199514a通过在传统处方中加入10~60%丙二醇的方法,使酮咯酸氨丁三醇注射液的澄明度得到了改善,使灭菌后的白色小点得到改善,但该制剂中均使用丙二醇溶剂,还是不能避免目前使用丙二醇带来的不良刺激,不利于药物的推广应用,且有关物质增长迅速。
6.中国专利cn102846542a通过在处方中加入甘油,改善注射液放置后的澄明度,还加入泊洛沙姆188,希望同时提高注射液的澄明度和放置稳定性。但由于选用了胆固醇、油酸、油酸钠、甘油或泊洛沙姆188作为稳定剂,胆固醇、油酸等易被氧化;泊洛沙姆作为一种表面活性剂,具有一定的溶血性和致敏性有溶血和致敏风险,容易引发安全性问题。
7.已上市制剂和公开文献中报道的药物活性成分,都是酮咯酸氨丁三醇,其酸根酮咯酸是外消旋体,由左酮咯酸和右酮咯酸等量构成。酮咯酸容易引发全身过敏、胃肠穿孔、胃出血、哮喘、肺水肿,肾功能衰竭等严重不良反应,这些严重的副作用限制了酮咯酸在疼痛治疗中的剂量和用药时间。左酮咯酸作为其有效单一异构体,能够减小无镇痛效果的右
酮咯酸带来的肾代谢负担,可能是减少不良反应的一种开发探索。
8.左酮咯酸的旋光值为负数,立体构型为s型对映体,结构如下式ⅰ化合物所示。左酮咯酸发挥主要的镇痛药效。右酮咯酸的旋光值为正数,立体构型为r型对映体。右酮咯酸几乎不发挥镇痛药效,可能是酮咯酸引发较多不良反应的原因之一,结构如下所示。
9.寻求和开发酮咯酸的单一异构体药物也越来越引发研究人员的关注。
[0010][0011]
研究表明,左酮咯酸的药效大大强于右酮咯酸。左酮咯酸镇痛药效是右酮咯酸的230倍,左酮咯酸的消炎药效是右酮咯酸的60倍。因此,如果采用单一的s
‑
异构体给药,则在产生相同药效的情况下,给药量可以比外消旋体给药降低近一半,从而降低了毒副作用。因此,获得光学活性的s
‑
异构体是有意义的。左酮咯酸在小鼠、大鼠体内,左、右酮咯酸之间发生不同程度的转化,而在人体内左酮咯酸向右酮咯酸转化的比例小于等于6.5%(参见文献j cln pharmacol 1996;36:521
‑
539)。单一的左酮咯酸的制剂具有降低剂量、疗效更佳、安全性更好的优点,具有广阔的开发前景。
[0012]
酮咯酸具有独特的化学结构,即吡咯并吡咯烷的杂环,不同于常见的芳基丙酸类非甾体抗炎药,比如布洛芬。左酮咯酸的手性中心位于这个特殊的吡咯并吡咯烷杂环上,导致该分子在温度升高尤其是溶液状态时极易发生消旋化,产生更多的异构体杂质右酮咯酸。
[0013]
中国专利cn108451909a报道,“酮咯酸氨丁三醇对光、热、酸、碱均不稳定,易发生脱羧氧化反应产生杂质,在一定程度上影响了临床用药安全”。gu等(international journal ofpharmaceutics,1988,41,105
‑
113)报道了,酮咯酸氨丁三醇在光照条件下通过光解自由基的机理生成了氧化态中间体(原文标注杂质2),再进一步生成1
‑
酮基酮咯酸(原文标注杂质3)和1
‑
羟基酮咯酸(原文标注杂质1)),相关化合物结构如下所示,
[0014][0015]
现有公开文献中尚未涉及如何在左酮咯酸的注射剂中保持光学稳定性的报道和提示。我们经过文献查阅和实验摸索,发现左酮咯酸的制剂较难保持其中原料药的手性光学纯度。因此,如何运用制剂技术手段能够保持左酮咯酸在制剂中的光学稳定性,是一项具有挑战性的课题,亟待解决。
技术实现要素:
[0016]
左酮咯酸是酮咯酸氨丁三醇中发挥药效的活性光学异构体,将其从外消旋体中单独分离出来制成左酮咯酸的制剂形式,有利于提高单位给药剂量的药效、降低酮咯酸氨丁三醇较为明显的不良反应,具有广阔的临床应用前景。
[0017]
鉴于:1)目前上市销售的酮咯酸制剂中的药物成份都是外消旋的酮咯酸,并没有单一立体异构体作为原料药的上市药物;2)左酮咯酸由于手性中心所在的特殊位置,在高温高湿条件下或者高温溶液状态下极易发生手性中心的消旋化;3)目前公开文献和资料尚未报道过左酮咯酸的注射液的制备以及保持光学稳定性的先例。
[0018]
因此,为解决现有技术中有关于左酮咯酸注射剂保持光学稳定性的技术空白和技术缺陷,本发明的目的在于提供一种左酮咯酸的药物组合物,该药物组合物中包括:
①
式ⅰ化合物即左酮咯酸或其药学上可接受的盐或溶剂合物;
②
ph调节剂;
③
药学上可接受的赋形剂。其中,左酮咯酸的化学结构如式ⅰ化合物所示,
[0019][0020]
本发明所述药物组合物,所述药学上可接受的盐选自:左酮咯酸钠盐、左酮咯酸氨丁三醇盐、左酮咯酸二乙胺盐、左酮咯酸乙二胺盐、左酮咯酸赖氨酸盐、左酮咯酸精氨酸盐、左酮咯酸组氨酸盐、左酮咯酸葡甲胺盐中的一种或几种。
[0021]
本发明所述药物组合物,在制剂形式上为:冻干粉针剂,或者溶液型注射剂,或者是将各组分均匀混合的无菌粉末。
[0022]
本发明所述的药物组合物,优选为冻干粉针剂;所述冻干粉针剂在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于90%;并且,该冻干粉针剂的水分含量小于等于5%,优选为小于等于3%,更加优选为小于等于2%。
[0023]
本发明优选实施方案中,制剂所用的原料药选自:左酮咯酸、左酮咯酸钠盐、左酮咯酸氨丁三醇盐、左酮咯酸二乙胺盐、左酮咯酸乙二胺盐、左酮咯酸赖氨酸盐、左酮咯酸精氨酸盐、左酮咯酸组氨酸盐、左酮咯酸葡甲胺盐;
[0024]
ph调节剂包含“缓冲剂”和“定容ph值调节剂”两部分,“缓冲剂”为无水或含结晶水的磷酸盐及其溶液、醋酸盐及其溶液、枸橼酸盐及其溶液、三乙胺缓冲溶液、硼砂缓冲液,或其混合物;优选磷酸盐及其溶液,或者醋酸盐及其溶液,或者枸橼酸盐及其溶液;其中,无水或含结晶水的磷酸盐选自磷酸二氢钠、磷酸氢二钠、磷酸二氢钾、磷酸氢二钾、磷酸钠、磷酸三钾、磷酸氢钠、磷酸氢钾、磷酸氢铵、磷酸二氢铵、磷酸铵、磷酸二氢钙、磷酸氢钙或磷酸锌中的一种或几种;
[0025]“定容ph值调节剂”选自氢氧化钠、氢氧化钾、氨丁三醇、三乙醇胺、二乙醇胺、乙醇胺、柠檬酸钠、柠檬酸钾、碳酸钠、碳酸氢钠、磷酸氢二钠的无水或结晶水合物、磷酸二氢钠的无水或结晶水合物、磷酸氢二钾的无水或结晶水合物、磷酸二氢钾的无水或结晶水合物、磷酸钠、磷酸、盐酸、酒石酸、乳酸;优选磷酸氢二钠的无水或结晶水合物、磷酸二氢钠的无水或结晶水合物、磷酸,或者其配制成的水溶液;
[0026]
赋形剂选自甘露醇、右旋糖酐、蔗糖、麦芽糖、乳糖、甘氨酸、麦芽糊精,或聚维酮k30;优选为甘露醇或麦芽糊精。本发明优选甘露醇或/和麦芽糊精作为冻干赋形剂,相较于
其他赋形剂,甘露醇或/和麦芽糊精的配伍最稳定,应用效果最好。
[0027]
本发明优选实施方案中,所述定容ph值调节剂用于调节该冻干粉针剂制备时的冻干原液的ph值为6.5~7.5,优选为6.8~7.2;更加优选为6.9~7.0。
[0028]
本发明优选实施方案中,该冻干粉针剂制备时的冻干原液中,以左酮咯酸c
15
h
13
no3计的药物浓度为2~30mg/ml,优选为5~10mg/ml。
[0029]
本发明所得的药物组合物还可使用稀释剂稀释后使用,优选地,该冻干粉针剂可用临床上常用的稀释剂稀释后使用;稀释剂选自5%葡萄糖注射液、10%葡萄糖注射液、葡萄糖氯化钠注射液、0.9%氯化钠注射液、复方氯化钠注射液、乳酸钠林格注射液、复方葡萄糖乳酸钠注射液、灭菌注射用水、木糖醇注射液、果糖注射液;并且,稀释后以左酮咯酸c
15
h
13
no3计的药物浓度为0.05~5mg/ml。
[0030]
本发明优选实施方案中,该冻干粉针剂中,原料药选自左酮咯酸,或者左酮咯酸氨丁三醇盐,或者左酮咯酸精氨酸盐;在ph调节剂中,缓冲剂选自无水或含结晶水的磷酸盐,可以是无水或结晶水的磷酸氢二钠、无水或结晶水的磷酸二氢钠中的一种或几种;定容ph值调节剂选自无水或结晶水的磷酸氢二钠、无水或结晶水的磷酸二氢钠、磷酸、氢氧化钠,或者其配制成的水溶液;赋形剂选自甘露醇;并且,该冻干粉针剂制备时的冻干原液中,以左酮咯酸计的药物浓度为5~7.5mg/ml;冻干原液在进行冷冻干燥前的ph值为6.8~7.2。
[0031]
本发明制剂所用的原料药,以左酮咯酸为例,其为类白色至淡黄色固体粉末,在水中几乎不溶或不溶;在二氯甲烷微溶;在无水乙醇略溶;在甲醇中溶解;在二甲基甲酰胺中易溶。
[0032]
通过理化试验探索,左酮咯酸的溶解度为ph依赖型,其溶解度随ph的提高而升高,酸性条件下ph值在6.0以下不利于活性成分左酮咯酸的溶解,制品成型性、复溶效果都不好,ph值在6.8以上具有更高的溶解度;ph在6.0~7.5的范围内时,随ph的升高,左酮咯酸发生构象翻转生成右酮咯酸的速率很低,显示左酮咯酸冻干制剂的光学稳定性更好;而当ph大于7.5时左酮咯酸发生构象翻转生成右酮咯酸的速率升高,冻干制剂中右酮咯酸含量增加,复溶溶液光学稳定性降低。
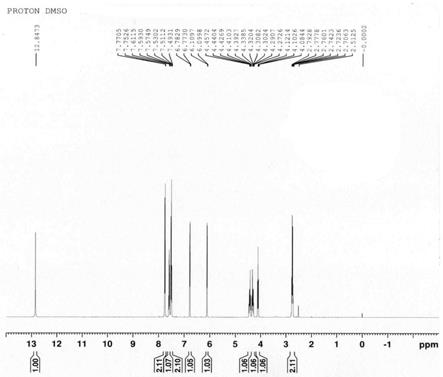
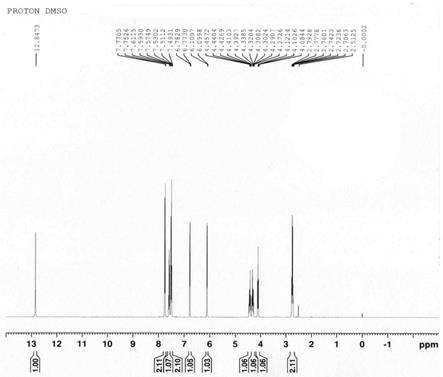
[0033]
本发明优选实施方案中,该冻干粉针剂中,作为ph调节剂使用的磷酸盐的摩尔浓度为0.01~0.1mmol/ml,优选为0.05~0.06mmol/ml;并且,冻干原液中左酮咯酸:磷酸盐的摩尔比为1:1~1:3,优选为1:2.5~1:2.8;赋形剂甘露醇在冻干原液中的质量百分数为3%~7%,优选5%。
[0034]
可见异物和不溶性微粒检查是冻干粉针剂的质量控制的重要检查项,直接关系到注射给药的用药安全性。这两项与冻干粉针的复溶的性质密切相关。经过处方筛选,结合复溶性质、水分控制等指标,我们惊奇地发现,通过加入葡甲胺、氨丁三醇、精氨酸等组分,有利于提高所得冻干粉针剂放置一段时间后复溶的质量,包括可见异物和不溶性微粒的检查指标都能稳定地保证合格。优选葡甲胺,或精氨酸。我们把此类组分在本发明中定义为“稳定剂”,用于防止冻干粉针剂复溶时的药物析出,有效改善可见异物和不溶性微粒的检测指标。回顾现有技术,大多使用乙醇、丙二醇等有机溶剂,作为助溶剂对药物成份酮咯酸进行助溶,可在一定程度上改善注射液澄明度问题,时灭菌后的白色小点得到改善,但有机醇类的使用不可避免带来给药时的较大刺激性,不利于药物的推广应用。
[0035]
本发明优选实施方案中,该冻干粉针剂中,还含有稳定剂,稳定剂选自葡甲胺、精
氨酸、赖氨酸、氨丁三醇中的一种或几种,优选为葡甲胺或精氨酸,稳定剂在冻干原液中的质量百分数为0.1%~5%,优选为0.5%~1%。
[0036]
酮咯酸在光照和/或高温下容易通过氧化态自由基中间体形式过度,脱羧氧化,进而生成1
‑
酮基酮咯酸和1
‑
羟基酮咯酸。通过添加抗氧剂例如亚硫酸氢钠等可以降低或防止生成氧化态自由基,从而较少脱羧氧化杂质产生。同时,氧化反应是稳定性中的很复杂的一类降解。氧化反应一般包括自由基的自氧化反应。氧化降解反应受到光照、金属离子(铜离子、铁离子)、温湿度、溶液或者固体影响较大。例如有些药物api固体不敏感,而溶液状态就很敏感。因此,通过加入适量少量的金属离子螯合剂例如依地酸二钠,可以削弱金属离子对氧化的催化加速作用,进而也能在一定程度上抑制杂质生成。
[0037]
本发明优选实施方案中,该冻干粉针剂中,还含有金属离子螯合剂、抗氧剂中的一种或两种;其中,金属离子螯合剂选自依地酸二钠、依地酸钙钠、枸橼酸钠、枸橼酸中的一种或几种,优选为依地酸二钠或依地酸钙钠,其在冻干原液中的重量百分数为0.05%~1%;抗氧剂选自硫代硫酸钠、亚硫酸钠、焦亚硫酸钠、亚硫酸氢钠、维生素c、枸橼酸钠、枸橼酸、l
‑
半胱氨酸中的一种或几种,优选为亚硫酸氢钠或维生素c,其在冻干原液中的重量百分数为0.02%~2%。
[0038]
本发明优选无水或结晶水的磷酸氢二钠、磷酸二氢钠、磷酸氢二钾、磷酸二氢钾作为ph调节剂,以维持溶解主药和保持光学稳定性两者之间的平衡。
[0039]
由于磷酸缓冲盐通常具有无水物和各种不同数目水合物之分,因此特作说明如下:本技术的发明内容和实施例中所提及的磷酸氢二钠,可以使用无水磷酸氢二钠,也可以使用磷酸氢二钠十二水合物,还可以使用磷酸氢二钠一水合物、磷酸氢二钠二水合物、磷酸氢二钠五水合物、磷酸氢二钠七水合物。如未特别指出,通常采用的是磷酸氢二钠十二水合物。同理,磷酸二氢钠可以使用无水磷酸二氢钠,或者磷酸二氢钠一水合物,或者磷酸二氢钠二水合物。以上试剂的应用,应当理解为,使用相同摩尔数、即相同的物质的量的无水物或者其他数目的水合物,均能达到相同的缓冲和调节ph值的效果。因为无水物或不同数目水合物的分子量不同,只是体现投料重量上略有差别。此外,磷酸氢二钾、磷酸二氢钾具有类似的情况和说明。其他试剂如有相似情况,以此类推,不在赘述。
[0040]
本发明优选实施方案中,该冻干粉针剂中,原料药是左酮咯酸;在ph调节剂中,缓冲剂为无水或结晶水的磷酸氢二钠;定容ph值调节剂为无水或结晶水的磷酸氢二钠,或者其配制成的水溶液;还含有稳定剂葡甲胺,稳定剂在冻干原液中的质量百分数为0.1%~5%,优选为0.5%~1%;并且,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于90%;更加优选地,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于98%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%。
[0041]
本发明优选实施方案中,该冻干粉针剂中,原料药是左酮咯酸;在ph调节剂中,缓冲剂为磷酸钠、磷酸二氢钠水合物;定容ph值调节剂为氢氧化钠、磷酸,或者其配制成的水溶液;还含有稳定剂精氨酸,稳定剂在冻干原液中的质量百分数为0.1%~5%,优选为0.5%~1%;并且,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下
放置6个月后,活性成分的光学纯度大于等于95%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于90%;更加优选地,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于98%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%。
[0042]
本发明优选实施方案中,该冻干粉针剂中,原料药是左酮咯酸氨丁三醇盐;在ph调节剂中,缓冲剂为无水或结晶水的磷酸氢二钾、无水或结晶水的磷酸二氢钾;定容ph值调节剂为碳酸氢钠、盐酸,或者其配制成的水溶液;还含有稳定剂精氨酸,稳定剂在冻干原液中的质量百分数为0.1%~5%,优选为0.5%~1%;并且,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于90%;更加优选地,该冻干粉针剂,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于98%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%
[0043]
本发明优选实施方案中,该冻干粉针剂中,还含有金属离子螯合剂,选自依地酸二钠或依地酸钙钠,其在冻干原液中的重量百分数为0.05%~1%。
[0044]
本发明优选实施方案中,该冻干粉针剂中,还含有抗氧剂,选自亚硫酸氢钠或维生素c,其在冻干原液中的重量百分数为0.02%~2%。
[0045]
作为本发明优选的实施方案,该冻干粉针剂制备时的冻干原液中各组分的投料重量百分含量如下所示,
[0046]
左酮咯酸:0.5%~0.7%
[0047]
磷酸氢二钠十二水合物:1.8%~2.0%
[0048]
甘露醇:6%~9%
[0049]
依地酸二钠:0.05%~0.1%
[0050]
磷酸氢二钠水溶液:适量,调节ph值为6.8~7.2;
[0051]
注射用水加至:100%;
[0052]
并且,将上述冻干原液进行无菌过滤、分装、冷冻干燥,即得该冻干粉针剂。
[0053]
作为本发明优选的实施方案,该冻干粉针剂制备时的冻干原液中各组分的投料重量百分含量如下所示,
[0054]
左酮咯酸:0.5%~1%
[0055]
磷酸氢二钠十二水合物:1.8%~2.5%
[0056]
甘露醇:5%~15%
[0057]
葡甲胺:1%~2%
[0058]
磷酸氢二钠水溶液:适量,调节ph值为6.8~7.2;
[0059]
注射用水加至:100%;
[0060]
并且,将上述冻干原液进行无菌过滤、分装、冷冻干燥,即得该冻干粉针剂。
[0061]
作为本发明优选的实施方案,该冻干粉针剂制备时的冻干原液中各组分的投料重量百分含量如下所示,
[0062]
左酮咯酸氨丁三醇盐:0.8%~1.5%
[0063]
磷酸二氢钾:0.5%~1.5%
[0064]
甘露醇:5%~10%
[0065]
精氨酸:1%~2%
[0066]
磷酸氢二钾水溶液,适量,调节ph值为6.8~7.2;
[0067]
注射用水加至:100%;
[0068]
并且,将上述冻干原液进行无菌过滤、分装、冷冻干燥,即得该冻干粉针剂。
[0069]
本发明目的之二是提供一种左酮咯酸的冻干粉针剂的制备方法,包括以下步骤:
[0070]
1)用70~80%的注射用水在室温下、搅拌条件下,溶解除了原料药、定容ph值调节剂之外的所有辅料,所述辅料是制缓冲剂、赋形剂、稳定剂、金属离子螯合剂、抗氧剂中的一种或几种,得到澄清溶液;
[0071]
2)加入左酮咯酸或其药学上可接受的盐,搅拌使其溶清;
[0072]
3)配制定容ph值调节剂,缓慢加入步骤2所得溶液中,调节ph至6.5~7.5,优选6.8~7.2,更加优选为6.9~7.0;
[0073]
4)用注射用水定容补足至总量,将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;
[0074]
5)将药液分装于中性硼硅玻璃管制注射剂瓶中,使用冷冻干燥设备执行冷冻干燥程序,最后压塞、出柜、轧盖,即得左酮咯酸冻干粉针剂成品。
[0075]
本发明的目的之三是提供一种左酮咯酸的冻干粉针剂在制备镇痛药物中的应用,该镇痛适应症具体为:需要阿片水平镇痛药的急性较严重疼痛的短期治疗,通常用于手术后镇痛。经评估判断,本发明所述的左酮咯酸冻干粉针剂可以缓解中度疼痛以及剧烈的手术之后的疼痛,包括腹部、胸部、妇科、口腔以及泌尿科等。也可以用于缓解肾绞痛、胆绞痛、牙齿疼痛、三叉神经痛以及癌性疼痛等。
[0076]
本发明首次报道了左酮咯酸或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐)的冻干粉针制剂及制备方法。该冻干粉针制剂在制备镇痛药物上具有重要用途,该镇痛适应症具体为:需要阿片水平镇痛药的急性较严重疼痛的短期治疗,通常用于手术后镇痛。
[0077]
本发明与现有技术相比,其显著优点是:
[0078]
(1)酮咯酸的镇痛活性来源于左酮咯酸,其对映异构体右酮咯酸几乎未发现镇痛作用。国内外市场销售的酮咯酸氨丁三醇是外消旋体,存在给药频繁、非甾体类药物共有的胃肠道不良反应、病人依从性差等缺陷;每日剂量为60~120mg,需多次给药维持药效。我们研究发现左酮咯酸可直接制备成冻干粉针剂,或者可将原料药先制成左酮咯酸的氨丁三醇盐、精氨酸盐、钠盐等盐型再形成冻干制剂,能够将传统给药剂量减小至约50%,具有性质稳定、高效起效、安全性高的优点。
[0079]
(2)本发明对活性成分左酮咯酸,或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐等)做了进一步的性质研究,发现其在水溶液中加剧降解和消旋化,右酮咯酸含量随着时间延长而增加,因此需严格控制外界条件以保证异构体杂质不超限度。我们发现将左酮咯酸直接制备成冻干粉针剂,有利于左酮咯酸本身保持光学纯度。我们通过制剂的处方和工艺的调整将异构体杂质右酮咯酸含量控制在质量限度范围内。我们采用冻干粉针剂的优化处方和工艺解决了左酮咯酸注射剂保持光学稳定性的技术难题和瓶颈。
[0080]
(3)在ph调节剂的筛选过程中,我们惊喜地发现,虽然左酮咯酸的水溶性较差,但是在与适当比例的磷酸缓冲盐共存条件下,经短时间搅拌,即可溶解得到澄清溶液,并且此时ph值测得为6.6~6.9,低于中性7.0。磷酸盐优选为无水或结晶水的磷酸盐,包含但不限于:磷酸氢二钠、磷酸二氢钠、磷酸氢二钾、磷酸二氢钾、磷酸钠。以上方案不仅提高左酮咯酸的水中溶解度和溶解速率,同时能够保证在溶解过程、制备冻干粉针的过程,以及成品在放置条件下的光学稳定性,对避免异构体杂质右酮咯酸的过度增长、减缓消旋化的速率,起到了积极作用。同时,该首次报道的技术方案避免了使用乙醇、丙二醇等有机溶剂作为助溶剂,避免了使用包合物例如环糊精或者非离子型表面活性剂例如泊洛沙姆188,减轻了注射剂应用时的血管、肌肉刺激性,降低了溶血等用药安全风险。本发明优选实施方案中,在长期稳定性试验即25℃
±
2℃/60%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于98%;在加速稳定性试验即40℃
±
2℃/75%rh
±
5%rh条件下放置6个月后,活性成分的光学纯度大于等于95%。
[0081]
(4)我们还探索了溶液ph值对主药稳定性的影响:酸性条件不利于活左酮咯酸的溶解,制剂成品的外观、均一性和复溶性能等指标都不佳。偏碱性条件下(ph大于7.5)会加剧左酮咯酸的降解和消旋化,右酮咯酸含量增加明显,因此确定最终适宜的ph在6.0~7.5间,更优选为6.8~7.2之间。该ph条件下的左酮咯酸或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐、左酮咯酸精氨酸盐)的理化稳定性质最佳。
[0082]
(5)我们对冻干工艺和成品水分控制做了深入研究。我们关注预冻和升华干燥这两个阶段,合理设置各阶段的温度和时间参数以保证冻干产品质量。改法制得的冻干成品,水份控制在≤2%。现有技术中,冻干粉针剂水份一般高于2%,通常为3%~5%。众所周知,由于酮咯酸本身遇到高温、光照和酸碱条件容易发生降解,颜色加深,其容易发生脱羧氧化反应产生杂质,一定程度上影响了临床用药安全。且溶液态下的降解速率明显比固体下快速。左酮咯酸不仅具有酮咯酸带来的化学不稳定性之外,光学稳定性也须十分关注,手性原子极易消旋。因此将左酮咯酸冻干制剂的水分控制在合理范围内,有利于增加样品放置过程中的化学稳定性和光学稳定性。
[0083]
(6)本发明所述左酮咯酸或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐、左酮咯酸精氨酸盐),其作为原料药,工艺稳定,操作方便,符合工业化大生产的要求。
[0084]
(7)本发明优选的实施方案中,加入了“稳定剂”,例如精氨酸或葡甲胺,可以有效改善冻干粉针剂放置一段时间后复溶溶液的可见异物和不溶性微粒等澄明度检测结果。
[0085]
(8)本发明优选的实施方案中,加入了金属离子螯合剂或抗氧剂,有助于降低药物成份在制剂制备和样品储存过程中的产生的脱羧氧化杂质,例如“1
‑
酮基酮咯酸”和“1
‑
羟基酮咯酸”等杂质,还能一定程度上抑制左酮咯酸的立体转化。
附图说明
[0086]
图1是左酮咯酸的核磁共振氢谱。
[0087]
图2是左酮咯酸的高分辨质谱。
[0088]
图3是制剂处方对异构体杂质右酮咯酸(%)的主效应示意图。
[0089]
图4是制剂处方对异构体杂质右酮咯酸(%)交互作用示意图。
[0090]
图5是本发明所述的冻干粉针剂在25℃下储存6个月的稳定性检测代表性图谱。
[0091]
图6是本发明所述的冻干粉针剂在40℃下储存6个月的稳定性检测代表性图谱。
具体实施方式
[0092]
为了使本公开的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施例对本公开的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本公开,但是本公开还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本公开内涵的情况下做类似推广,因此本公开不受下面公开的具体实施例的限制。
[0093]
试验例1左酮咯酸的制备和结构确证
[0094]
现有文献中已有涉及关于左酮咯酸的制备。fulling等(j.am.chem.soc.,1987,109,2845)通过用酶选择性催化水解酮咯酸酯获得s
‑
酮咯酸的方法,产品具有高的光学纯度。guzman等(j.med.chem.,1986,29,589
‑
591)用l
‑
辛可尼丁(l
‑
cinchonidine)与酮咯酸反应生成盐,通过结晶方法拆分了酮咯酸。angel等(tetrahydron:asymmetry,1992,3(11),1455)使酮咯酸与光学活性醇反应生成酯,经高压液相色谱(hplc)分离后水解,获得( )
‑
或(
‑
)
‑
酮咯酸的方法。
[0095]
本发明使用辛可尼丁拆分的方法,获得了左酮咯酸样品。制备过程描述如下:
[0096]
将11.7g的l
‑
辛可尼丁溶于200ml热的无水乙醇中,加入到10.2g外消旋酮咯酸在50ml乙酸乙酯的溶液中。此混合物加热至回流的温度反应0.5h,减压去除溶剂,残余物用200ml乙酸乙酯重结晶,得到约3.7g的左酮咯酸。
[0097]
1)比旋度为
‑
176
°
至
‑
177
°
(c=0.1,乙醇)。
[0098]
2)核磁氢谱数据如下,参见附图1。
[0099]
[0100][0101]
3)高分辨质谱数据如下,参见附图2。
[0102][0103]
正离子模式下([c
15
h
13
no3] h)
的m/z为256.09692,说明测试样品的质荷比为255.089808,经计算得分子组成为c
15
h
13
no3。符合目标分子结构。
[0104]
由于左酮咯酸本身的水溶性较差,水溶液在上高温或光照下容易降解变色,使得有关物质升高和异构体杂质右酮咯酸增加。因此,我们考察了将左酮咯酸制备成常见的盐型,例如左酮咯酸钠盐、左酮咯酸精氨酸盐。制备常规盐型的方法概括描述为将左酮咯酸与等摩尔量的碱基,溶于无水乙醇中,室温下搅拌0.5至2h,随后减压去除溶剂,加入乙酸乙酯和/或正己烷,所得固体沉淀,抽滤,减压干燥,即得盐型成品。
[0105]
试验例2左酮咯酸在不同ph磷酸盐缓冲液中的溶解度试验
[0106]
将左酮咯酸进一步制备成左酮咯酸氨丁三醇盐、左酮咯酸钠盐后,原料药固体状态下的稳定性有提升,在高温、高湿、强光照射下,稳定性显著提高,外观始终为白色或类白色,无颜色加深现象。但在水溶液中,高温条件下仍然会加剧其降解和消旋化,即右酮咯酸的含量会显著增加。
[0107]
左酮咯酸在水溶液中的溶解度不佳,在筛选ph调节剂的过程中,我们惊喜地发现在左酮咯酸的水溶液中加入磷酸盐即磷酸氢二钠或/和磷酸二氢钠,不仅有助于提高主药酮咯酸在水中的溶解度,而且能够大大提高左酮咯酸在制剂中的光学稳定性,减缓其在加速稳定性试验条件下的消旋化速率。
[0108]
参考中国药典2020配制ph范围在6.5
‑
7.8的磷酸盐缓冲液,分别加入适量左酮咯酸(游离酸)制成约含20mg/ml的混合物,将各溶液置于25℃空气浴摇床振摇24小时,期间观
察各混合物中左酮咯酸的溶解情况,检测各ph条件下化合物在25℃、1天后的溶解度,如表1所示,剩余样品置25℃和40℃条件下留样,于2天、30天取样检测异构体的生成情况,如表2所示。
[0109]
表1左酮咯酸在不同ph值磷酸缓冲液汇中的溶解度
[0110][0111][0112]
表2左酮咯酸在不同ph磷酸盐缓冲液中的光学稳定性
[0113][0114]
上述试验结果表明:1)左酮咯酸为ph依赖性溶解,其溶解度随酸度值的提高而升高,在ph为7.8的磷酸盐缓冲液中,溶解度发生突跃。2)样品的溶液状态下,在25℃条件下放置1个月,左酮咯酸的光学纯度大于等于95%,但40℃条件下放置1个月,异构体右酮咯酸生成显著增加,说明高温条件下左酮咯酸在磷酸盐缓冲溶液中仍然会加剧其降解和消旋化,光学稳定性有待通过技术手段来提高。
[0115]
综上可知,左酮咯酸或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐)很可能不适宜制备成常规的注射液水溶液以保持其光学纯度,有待进一步研究开发其适宜的剂型解决上述技术难题。冻干粉针剂与注射液水针相比有望保持药物的光学纯度,因为其在真空状态下制备不易被氧化,且含水量很低,被水催化产生的降解几率较低;且冻干粉针特殊的生产工艺使药品均匀分散在冻干保护剂中,具备稳定的制剂环境。综上,左酮咯酸,或其药学上可接受的盐(包括左酮咯酸氨丁三醇盐、左酮咯酸钠盐)适合开发成冻干粉针剂,以解决活性成分左酮咯酸注射水溶液难以保持其光学纯度、理化稳定性的难题。
[0116]
试验例3制剂中的赋形剂和冻干原液的ph值的筛选和考察
[0117]
制备方法:用70~80%的注射用水溶解赋形剂、ph调节剂磷酸二氢钠十二水合物,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用磷酸氢二钠溶液调节ph;用注射用水定容补足至总量;无菌过滤后,经冷冻干燥程序,即得左酮咯酸的冻干粉针剂成品。
[0118]
在25℃和40℃的温度条件下放置20天,考察不同赋形剂、不同冻干原液ph值的条件下,所制得冻干粉针剂中右酮咯酸含量的变化,结果见表4。
[0119]
表3不同赋形剂和不同冻干原液ph值的影响
[0120][0121]
表4不同赋形剂所得成品的性状
[0122][0123][0124]
上述试验结果表明:从冻干制剂成品在不同温度留样后的性状角度,以甘露醇、麦芽糊精和聚维酮k30为赋形剂的冻干样品符合要求。从不同留样条件下的异构体检测结果分析,以蔗糖、麦芽糖、麦芽糊精和甘露醇为赋形剂的冻干样品符合要求。综上,优选的赋形剂为麦芽糊精和甘露醇。
[0125]
试验例4冻干粉针剂用生理盐水稀释后的光学稳定性考察
[0126]
表5生理盐水稀释后的右酮咯酸考察
[0127][0128]
上述试验结果表明:各左酮咯酸冻干制剂在25℃留样24小时异构体均无明显增长,在40℃下留样24小时异构体稍有增长,仍在可控的质量范围内。
[0129]
试验例5左酮咯酸游离酸和不同盐型制成冻干粉针剂后的稳定性考察
[0130]
制备方法:与试验例3的制备方法类似。其中,ph调节剂为磷酸氢二钠或/和磷酸二氢钠,加入量随主药左酮咯酸或其药学上可接受的盐的含量而变化,浓度为3~7.5mg/ml,更优选为5g/ml。在加速试验(温度40
±
2℃,相对湿度75
±
5℃)的条件下放置6个月,考察样品的含量、右酮咯酸含量、外观、性状、复溶等各项指标的变化,结果如下。
[0131]
表6不同原料药制成冻干粉针剂的光学稳定性考察
[0132][0133]
上述试验结果表明:相对比处方1不添加ph调节剂,处方2~6在添加磷酸氢二钠或/和磷酸二氢钠后,降低了左酮咯酸的消旋化。
[0134]
试验例6甘露醇和不同ph值条件下所得样品的稳定性考察
[0135]
制备方法:与试验例3的制备方法类似。样品在室温(25℃
±
2℃)及加速试验(温度40
±
2℃,相对湿度75
±
5℃)的条件下放置15天,考察样品的右酮咯酸含量、性状、复溶速度等指标的变化,结果见表7。
[0136]
表7不同ph条件下制得样品的性质考察
[0137][0138][0139]
上述试验结果表明:ph在6.0~7.5的范围内,酸性条件下不利于活性成分左酮咯酸的溶解,制品成型性、复溶效果都不好。随ph的升高,左酮咯酸发生构象翻转生成右酮咯酸的速率很低,显示左酮咯酸冻干制剂的光学稳定性更好;而当ph大于7.5时左酮咯酸发生构象翻转生成右酮咯酸的速率升高,冻干制剂中右酮咯酸含量增加,复溶溶液光学稳定性
降低,且ph为7.8时冻干制剂复溶速度慢,需借助涡旋方可溶解,不便于临床使用。
[0140]
试验例7冻干工艺的探索
[0141]
我们对冷冻控制(即预冻)和升华干燥阶段,分阶段地考察了各阶段中不同温度和时间下样品的外观、性状、复溶、含水量等各项指标的变化,结果见表8。
[0142]
表8冻干参数的探索考察
[0143][0144]
上述试验结果表明:按照冻干工艺1制备的样品性状较差、含水量高,按照冻干工艺2~5制备的样品性状较好、含水量低。因此,确定最优的冻干工艺为:半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,冻干结束。
[0145]
试验例8稳定剂的效果考察
[0146][0147]
称取处方量注射用水,于室温下搅拌,加入处方量葡甲胺和甘露醇,待溶解完全后加入处方量左酮咯酸,测量药液酸度值,并用磷酸氢二钠或磷酸将溶液ph调节至6.9
‑
7.1。将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量检测。将药液分装于5ml中性硼硅玻璃管制注射剂瓶中,装量依照中间体含量确
定,规格为每瓶含左酮咯酸10mg。
[0148]
冻干程序如下:
[0149]
a.预冻:于20分钟内降温至
‑
5℃并保温1小时;于20分钟内降温至
‑
10℃保温1h;于20分钟内降温至
‑
30℃并于保温6小时,预冻结束。
[0150]
b.升华干燥:将冻干机内真空度调节至20pa以下进行真空干燥。于20分钟内升温至
‑
20℃并保温9h;于20分钟内升温至
‑
10℃并保温8h;于20分钟内升温至0℃并保温8小时,升华干燥结束。
[0151]
c.解析干燥:将冻干机内真空度保持在20pa以下,于20分钟内升温至10℃并保温6h;将冻干机内真空度调节至10pa以下,于20分钟内升温至20℃并保温6h;将冻干机内真空度保持在10pa以下,于20分钟内升温至升温至30℃并保温3h,解析干燥结束。
[0152]
d.压塞、出柜,冻干结束。
[0153]
一、从复溶后制剂可见异物和不溶性微粒角度考察
[0154]
取各实施例样品若干,分别于5℃、25℃、40℃恒温恒湿箱中,留样6个月后,于各留样条件下取出各实施例样品2瓶。于室温静置下加入生理盐水2ml以复溶,记录瓶中块状物完全溶解所需时间;复溶后的溶液,参照中国药典2020版通则0904项进行可见异物灯检法检查,若无肉眼可见异物则参照中国药典2020版通则0903项进行不溶性微粒的显微镜观察,并记录如下:
[0155][0156]
上述试验结果表明:处方中加入适量的葡甲胺(≥0.5%),有益于制剂的复溶,有利于提高制剂在临床使用的稳定性和安全性。
[0157]
二、长期留样的异构体变化考察
[0158]
取各实施例样品若干,分别于5℃、25℃、40℃恒温恒湿箱中,分别于1个月、3个月、6个月,从各留样条件下取出各实施例样品1瓶进行异构体检测,左酮咯酸含量记录如下。
[0159][0160]
上述试验结果表明:各实施例于留样条件下,左酮咯酸的消旋化随着葡甲胺处方用量的增加而增加,从原料化合物光学稳定的角度考虑,葡甲胺的优选用量应≤1%。
[0161]
下述实施例1~12进一步描述本发明,且均能实现上述试验例所述的效果,但所述的实施例仅用于说明本发明而不是限制本发明。
[0162]
实施例1左酮咯酸钠盐的冻干粉针剂的制备
[0163]
处方:左酮咯酸钠盐:1.1g;甘露醇:11g;无水磷酸氢二钠:1.44g;5%磷酸二氢钠溶液:适量,调至ph6.8;注射用水:补至总量200g。
[0164]
制备方法:
[0165]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、无水磷酸氢二钠,得澄清溶液,再加入左酮咯酸钠盐搅拌使其溶清,用5%磷酸二氢钠溶液调节ph至6.8;用注射用水定容补足至总量;
[0166]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸钠盐的冻干粉针剂成品。其相关检测数据如下。
[0167]
[0168]
实施例2左酮咯酸氨丁三醇盐冻干粉针剂的制备
[0169]
处方:左酮咯酸氨丁三醇盐:1.5g;甘露醇:10g;磷酸二氢钠十二水合物:1.44g;0.5mol/l磷酸二氢钠溶液:适量,调至ph6.5;注射用水:补至总量200g。
[0170]
制备方法:
[0171]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物,得澄清溶液,再加入左酮咯酸氨丁三醇盐搅拌使其溶清,用0.5mol/l磷酸二氢钠溶液调节ph至6.5;用注射用水定容补足至总量;
[0172]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸氨丁三醇盐的冻干粉针剂成品。其相关检测数据如下。
[0173][0174]
实施例3左酮咯酸精氨酸盐冻干粉针剂的制备
[0175]
处方:左酮咯酸精氨酸盐:1.7g;甘露醇:14g;柠檬酸钠:1.5g;0.5mol/l柠檬酸溶液:适量,调至ph7.0;注射用水:补至总量200g。
[0176]
制备方法:
[0177]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、柠檬酸钠,得澄清溶液,再加入左酮咯酸精氨酸盐搅拌使其溶清,用0.5mol/l柠檬酸溶液调节ph至7.0;用注射用水定容补足至总量;
[0178]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸精氨酸盐的冻干粉针剂成品。其相关检测数据如下。
[0179][0180]
实施例4左酮咯酸冻干粉针剂的制备
[0181]
处方:左酮咯酸:1.0g;甘露醇:10g;依地酸二钠:0.1g;磷酸二氢钠十二水合物:3.60g;0.5mol/l磷酸氢二钠溶液:适量,调至ph7.2;注射用水:补至总量200g。
[0182]
制备方法:
[0183]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、依地酸二钠,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.2;用注射用水定容补足至总量;
[0184]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂成品。其相关检测数据如下:
[0185][0186][0187]
实施例5左酮咯酸冻干粉针剂的制备
[0188]
处方:左酮咯酸:1.0g;甘露醇:10g;葡甲胺:2.0g;磷酸二氢钠十二水合物:3.64g;0.5mol/l磷酸氢二钠溶液:适量,调至ph7.0;注射用水:补至总量200g。
[0189]
制备方法:
[0190]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、葡甲胺,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.0;用注射用水定容补足至总量;
[0191]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂成品。其相关检测数据如下。
[0192]
时间点性状右酮咯酸(%)复溶溶液可见异物及不溶性微粒检查0天白色块状物0.42符合要求5℃、1月白色块状物0.42符合要求
5℃、2月白色块状物0.42符合要求5℃、3月白色块状物0.43符合要求5℃、6月白色块状物0.43符合要求25℃、1月白色块状物0.41符合要求25℃、2月白色块状物0.46符合要求25℃、3月白色块状物0.48符合要求25℃、6月白色块状物0.58符合要求40℃、1月白色块状物0.73符合要求40℃、2月白色块状物0.91符合要求40℃、3月白色块状物1.00符合要求40℃、6月白色块状物1.32符合要求
[0193]
实施例6左酮咯酸氨丁三醇盐冻干粉针剂的制备
[0194]
处方:左酮咯酸氨丁三醇盐:3.0g;甘露醇:15g;依地酸钙钠:0.2g;醋酸钠:3.64g;0.5mol/l醋酸溶液:适量,调至ph6.9;注射用水:补至总量200g。
[0195]
制备方法:
[0196]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、葡甲胺,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.0;用注射用水定容补足至总量;
[0197]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂成品。其相关检测数据如下。
[0198]
时间点性状右酮咯酸(%)复溶溶液可见异物及不溶性微粒检查0天白色块状物0.53符合要求5℃、1月白色块状物0.53符合要求5℃、2月白色块状物0.53符合要求5℃、3月白色块状物0.55符合要求5℃、6月白色块状物0.56符合要求25℃、1月白色块状物0.58符合要求25℃、2月白色块状物0.61符合要求25℃、3月白色块状物0.69符合要求25℃、6月白色块状物0.75符合要求40℃、1月白色块状物0.72符合要求40℃、2月白色块状物0.96符合要求40℃、3月白色块状物1.05符合要求40℃、6月白色块状物1.43符合要求
[0199]
实施例7左酮咯酸精氨酸盐冻干粉针剂的制备
[0200]
处方:左酮咯酸精氨酸盐:2.0g;麦芽糊精:13g;亚硫酸氢钠:0.2g;磷酸钠:3.1g;
0.5mol/l磷酸溶液:适量,调至ph7.1;注射用水:补至总量200g。
[0201]
制备方法:
[0202]
步骤1:按上述处方,用70~80%的注射用水溶解麦芽糊精、磷酸钠、亚硫酸氢钠,得澄清溶液,再加入左酮咯酸精氨酸盐搅拌使其溶清,用0.5mol/l磷酸溶液调节ph至7.0;用注射用水定容补足至总量;
[0203]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸精氨酸的冻干粉针剂成品。其相关检测数据如下。
[0204]
时间点性状右酮咯酸(%)复溶溶液可见异物及不溶性微粒检查0天白色块状物0.67符合要求5℃、1月白色块状物0.67符合要求5℃、2月白色块状物0.68符合要求5℃、3月白色块状物0.69符合要求5℃、6月白色块状物0.72符合要求25℃、1月白色块状物0.69符合要求25℃、2月白色块状物0.73符合要求25℃、3月白色块状物0.80符合要求25℃、6月白色块状物0.88符合要求40℃、1月白色块状物0.73符合要求40℃、2月白色块状物0.96符合要求40℃、3月白色块状物1.2符合要求40℃、6月白色块状物1.62符合要求
[0205]
实施例8左酮咯酸冻干粉针剂的制备
[0206]
处方:左酮咯酸:1.0g;甘露醇:10g;葡甲胺:1.5g;维生素c:0.5g;磷酸二氢钠十二水合物:3.64g;0.5mol/l磷酸氢二钠溶液:适量,调至ph7.0;注射用水:补至总量200g。
[0207]
制备方法:
[0208]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、葡甲胺、维生素c,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.0;用注射用水定容补足至总量;
[0209]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂成品。其相关检测数据如下。
[0210][0211]
实施例9左酮咯酸氨丁三醇盐冻干粉针剂的制备
[0212]
处方:左酮咯酸氨丁三醇盐:1.8g;甘露醇:12g;精氨酸:0.9g;l
‑
半胱氨酸:0.2g;柠檬酸:0.8g;0.5mol/l柠檬酸钠溶液:适量,调至ph7.2;注射用水:补至总量200g。制备方法:
[0213]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、精氨酸、l
‑
半胱氨酸、柠檬酸,得澄清溶液,再加入左酮咯酸氨丁三醇盐搅拌使其溶清,用0.5mol/l柠檬酸钠溶液调节ph至7.2;用注射用水定容补足至总量;
[0214]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸氨丁三醇盐的冻干粉针剂成品。其相关检测数据如下。
[0215][0216]
实施例10左酮咯酸冻干粉针剂的制备
[0217]
处方:左酮咯酸:2.0g;甘露醇:20g;葡甲胺:2.0g;磷酸二氢钠十二水合物:7.22g;0.5mol/l磷酸氢二钠溶液:适量,调至ph6.9~7.0;注射用水:补至总量400g。
[0218]
制备方法:
[0219]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、葡甲胺,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.0;用注射用水定容补足至总量;
[0220]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂
成品。
[0221]
实施例11左酮咯酸冻干粉针剂的制备
[0222]
处方:左酮咯酸:2.0g;甘露醇:20g;精氨酸:2.0g;磷酸二氢钠十二水合物:7.23g;0.5mol/l磷酸氢二钠溶液:适量,调至ph7.0~7.1;注射用水:补至总量400g。
[0223]
制备方法:
[0224]
步骤1:按上述处方,用70~80%的注射用水溶解甘露醇、磷酸二氢钠十二水合物、精氨酸,得澄清溶液,再加入左酮咯酸搅拌使其溶清,用0.5mol/l磷酸氢二钠溶液调节ph至7.0;用注射用水定容补足至总量;
[0225]
步骤2):将药液依次经过0.45μm、0.22μm、0.22μm孔径聚醚砜材质滤芯进行无菌过滤,对中间品溶液进行含量的质量控制;将药液分装于中性硼硅玻璃管制注射剂瓶中;半压加塞后,
‑
5℃~
‑
10℃预冻2h后冷却到
‑
40℃保温3h后,升温至
‑
20℃~0℃升华干燥8~12h,再升温至10~20℃,解析干燥6h~12h后压塞、出柜,最后轧盖,即得左酮咯酸的冻干粉针剂成品。
[0226]
实施例12比格犬和食蟹猴单次给药的药代动力学试验
[0227]
本发明所述的左酮咯酸冻干粉针剂,选用代表性批次进行了比格犬和食蟹猴的体内药物代谢试验考察。给药方式为单次静脉给药,采用液质联用方法(lc
‑
ms/ms)测定血浆样品中左酮咯酸和右酮咯酸的浓度,绘制血药浓度
‑
时间曲线,获取主要的药代动力学参数,考察两者在动物体内的手性转化情况。现将数据汇总如下。
[0228]
雌雄比格犬单次静脉给予不同剂量的左酮咯酸后药代动力学参数(平均值
±
标准偏差)
[0229][0230]
手性转化率(%)=(auc0‑
t
)
r
/[(auc0‑
t
)
s
(auc0‑
t
)
r
]
×
100
[0231]
na:无法获得。
[0232]
雌雄食蟹猴单次静脉给予不同剂量的左酮咯酸氨丁三醇后药代动力学参数(平均值
±
标准偏差)
[0233][0234]
手性转化率(%)=(auc0‑
t
)
r
/[(auc0‑
t
)
s
(auc0‑
t
)
r
]
×
100
[0235]
由上可知,在犬和猴体内,左酮咯酸向右酮咯酸的转化率较低,与文献(j cln pharmacol 1996;36:521
‑
539)报道的人体转化率6.5%接近。高剂量条件下左酮咯酸向消旋化的转化率没有明显地增加,这说明本发明所述的左酮咯酸冻干粉针剂经静脉注射入动物体内后,仍能够保持较高的左酮咯酸的占比,为高效地发挥镇痛药效奠定了良好基础。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。