1.本发明涉及包含新型离聚物的聚酰胺树脂组合物。
背景技术:
2.聚酰胺树脂由于机械特性、耐热性、电特性、成型加工性等优异,因此被广泛用于汽车部件、外装部件、机械部件、电气电子部件等。这些部件被成型为各种各样的形状并在各种用途和环境中使用,但聚酰胺树脂本质上存在耐冲击性低、在需要韧性的用途中难以施展的问题。
3.作为改善聚酰胺树脂的耐冲击性的尝试,对使聚酰胺树脂含有离聚物树脂作为改性剂进行了研究。例如日本特公昭55
‑
41659号公报提出了聚酰胺50~95重量%与乙烯系离聚物树脂和/或羧基改性丁腈橡胶5~50重量%的紧密混合物。另外,作为其它例子,日本特开2015
‑
163692号公报提出了相对于分子间和/或分子内具有基于金属离子的交联结构的聚烯烃系热塑性树脂100重量份,含有5~100重量份具有特定熔点的聚酰胺树脂的热塑性树脂组合物。日本特开2007
‑
204674号公报中,在特定聚酰胺中配混一定比例的离聚物来提高聚酰胺的成型性。列举的这些专利文献中,以聚酰胺树脂的耐冲击性等物性的改善为目的,使用了以往通常市售的离聚物。
4.离聚物为通过金属离子使高分子聚集而成的合成树脂,已知有作为以乙烯
‑
不饱和羧酸共聚物为前体树脂并通过钠、锌等金属离子进行分子间键合而成的树脂的乙烯系离聚物(美国专利第3264272号说明书)。乙烯系离聚物强韧并富有弹性,且具有柔软性,并且具有耐磨损性、透明性等特点。
5.目前,作为市售的乙烯系离聚物,已知有dupont公司开发的乙烯
‑
甲基丙烯酸共聚物的钠盐、锌盐“surlyn(注册商标)”、以及dow
‑
mitsui polychemicals co.,ltd.销售的“himilan(注册商标)”等。
6.但是,这些市售的乙烯系离聚物中使用的前体树脂的乙烯
‑
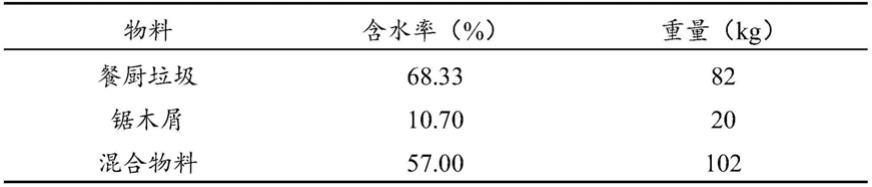
不饱和羧酸共聚物均使用了通过高压自由基聚合法使乙烯与不饱和羧酸等含极性基团的单体聚合而得到的含极性基团的烯烃共聚物。高压自由基聚合法的优点是相对而言不论含极性基团的单体的种类均可廉价地聚合。但是,如图1的形象图所示,由该高压自由基聚合法制造的含极性基团的烯烃共聚物的分子结构为不规则地具有多个长支链及短支链的结构,存在强度不充分的缺点。
7.为了改善由高压自由基聚合法得到的含极性基团的烯烃共聚物的缺点,摸索了以下方法:使用催化剂聚合出如图2的形象图所示的分子结构为直链状的含极性基团的烯烃共聚物。但是,含极性基团的单体通常会变为催化剂毒物,因此难以使用催化剂来聚合。因此,难以在工业上通过廉价且稳定的方法来得到具有期望的物性的含极性基团的烯烃共聚物。
8.但是近年来,提出了通过使用本技术人等研发的新催化剂及新制造方法从而在工业上廉价且稳定地获得分子结构基本上为直链状的含极性基团的烯烃共聚物的方法。
9.并且,对于作为乙烯系离聚物的前体树脂的含极性基团的烯烃共聚物的制造方法,本技术人等报告了使用后元素周期表的过渡金属催化剂,制造乙烯与丙烯酸叔丁酯的共聚物,通过对所得含极性基团的烯烃共聚物进行热或酸处理而将其改性为乙烯
‑
丙烯酸共聚物后,与金属离子进行反应,成功地制造了二元离聚物(日本特开2016
‑
79408号公报)。
10.现有技术文献
11.专利文献
12.专利文献1:日本特公昭55
‑
41659号公报
13.专利文献2:日本特开2015
‑
163692号公报
14.专利文献3:日本特开2007
‑
204674号公报
15.专利文献4:美国专利第3264272号说明书
16.专利文献5:日本特开2016
‑
79408号公报
技术实现要素:
17.发明要解决的问题
18.对聚酰胺的改性进行了各种尝试,但日本特公昭55
‑
41659号公报、日本特开2015
‑
163692号公报、日本特开2007
‑
204674号公报所述的离聚物是通过高压自由基聚合法制造的并且为不规则地具有多个长支链及短支链的结构,因此强度不充分。作为改性剂的离聚物本身具有强度的问题,在聚酰胺树脂组合物的耐冲击性改善方面不充分。另外,特别是日本特开2007
‑
204674号公报中仅针对特定的配混中的聚酰胺发现了能够起到效果,从赋予基于离聚物的物性的观点出发是不充分的。
19.鉴于相关以往技术的情况,本发明的目的在于提供能够提供耐冲击性优异的聚酰胺树脂的改性剂、以及通过配混该改性剂使得耐冲击性得到改善的聚酰胺树脂组合物。
20.用于解决问题的方案
21.为解决上述技术问题,本发明人等进行了深入研究,结果发现通过使用特定的离聚物树脂,对于包含聚酰胺的树脂的耐冲击性具有优异的效果。
22.日本特开2016
‑
79408号公报中记载的那样的乙烯系离聚物为以往没有的新型乙烯系离聚物,其前体树脂具有基本上为直链状的分子结构,同时还具有作为离聚物的功能,其物性等与以往的乙烯系离聚物大不相同,其特有的特性及适用的用途也是未知的。本发明是基于基本上为直链状的乙烯系离聚物出乎意料地在聚酰胺树脂的耐冲击性改善方面具有优异的效果这一发现而完成的。
23.即,本发明的第一方案为一种聚酰胺树脂组合物,其特征在于,其为包含聚酰胺树脂(i)和离聚物(ii)的聚酰胺树脂组合物,离聚物(ii)满足下述条件:包含源自乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)和源自具有羧基和/或二羧酸酐基团的单体的结构单元(b)作为必要构成单元的共聚物(p)中的、羧基和/或二羧酸酐基团的至少一部分被转换为含有选自元素周期表第1族、2族或12族中的至少1种金属离子的含金属羧酸盐,用旋转式流变仪测定的复数模量的绝对值g
*
=0.1mpa下的相位角δ为50度~75度。
24.发明的效果
25.根据本发明,可得到具备优异的耐冲击性的聚酰胺树脂组合物。包含基本上为直链状结构的本发明离聚物的本发明的聚酰胺树脂组合物与以往的聚酰胺树脂组合物相比,
耐冲击性优异,因此作为需要韧性的材料是有用的。
附图说明
26.图1为示出通过高压自由基聚合法制造的含极性基团的烯烃共聚物的分子结构的示意图。
27.图2为示出通过基于催化剂的聚合而得到的含极性基团的烯烃共聚物的分子结构的示意图。
具体实施方式
28.本发明为一种使用离聚物作为改性剂而得到的聚酰胺树脂组合物,该离聚物的特征在于:以包含源自乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)和源自具有羧基和/或二羧酸酐基团的单体的结构单元(b)作为必要构成单元并且这些结构单元基本上直链状共聚、优选无规共聚而得到的共聚物(p)作为前体树脂,该结构单元(b)的羧基和/或二羧酸酐基团的至少一部分被转换为含有选自元素周期表第1族、2族或12族中的至少1种金属离子的含金属羧酸盐。
29.下面,对本发明的离聚物、包含离聚物的聚酰胺树脂组合物及其用途等分别进行详细说明。需要说明的是,本说明书中,“(甲基)丙烯酸”是指丙烯酸或甲基丙烯酸。另外,本说明书中表示数值范围的“~”是指包含其前后记载的数值作为下限值和上限值。
30.1.聚酰胺树脂(i)
31.聚酰胺树脂为例如可通过3元环以上的内酰胺的开环聚合、氨基羧酸的缩聚或二羧酸与二胺的缩聚得到的分子链中具有酰胺键的高分子。作为开环聚合中使用的内酰胺的具体例子,可列举出ε
‑
己内酰胺、庚内酰胺、月桂内酰胺等。作为缩聚中使用的氨基羧酸的具体例子,可列举出6
‑
氨基己酸、7
‑
氨基庚酸、8
‑
氨基辛酸、9
‑
氨基壬酸、11
‑
氨基十一酸、12
‑
氨基十二酸等。作为二羧酸与二胺的缩聚中使用的单体的具体例子,可列举出1,4
‑
丁二胺、1,5
‑
戊二胺、1,6
‑
己二胺、1,5
‑
己二胺、1,9
‑
壬二胺、1,11
‑
十一烷二胺、1,12
‑
十二烷二胺、α,ω
‑
二氨基聚丙二醇、间或对苯二胺等二胺与己二酸、癸二酸、十二烷二酸、戊二酸、对苯二甲酸、间苯二甲酸等二羧酸的任意组合。聚酰胺树脂可以为由多种不同的单体得到的共聚物,也可以将均聚物或共聚物以任意比例共混来使用。
32.作为聚酰胺树脂,可以使用市售级的聚酰胺树脂,但作为更优选的聚酰胺的具体例子,可列举出6
‑
尼龙、6,6
‑
尼龙、6,10
‑
尼龙、12
‑
尼龙、11
‑
尼龙、9
‑
尼龙、7
‑
尼龙、聚酰胺4,6、聚酰胺6,12、聚间二甲苯己二酰胺、芳香族尼龙例如聚酰胺6t、聚酰胺9t、聚酰胺10t等。这些聚酰胺树脂可以单独使用,或者2种以上组合使用。
33.这些聚酰胺树脂的末端可以用碳数6~22的羧酸或胺封端。作为具体用于封端的羧酸,可列举出己酸、辛酸、癸酸、月桂酸、肉豆蔻酸、棕榈酸、硬脂酸、山萮酸等脂肪族单羧酸。另外,作为胺,可列举出己胺、辛胺、癸胺、月桂胺、肉豆蔻胺、棕榈胺、硬脂胺、山嵛胺等脂肪族伯胺。
34.聚酰胺树脂可根据具体用途在一定程度上自由调整,但优选具有一定范围内的分子量。高分子还可以用相对粘度(ηr)来表示分子链的长度,即分子量。聚酰胺树脂的优选相对粘度以按照jis k6810在98%硫酸中、树脂浓度1%、温度25℃下测定的值计为1.5~5.0
的范围,更优选为2.0~5.0的范围。
35.树脂组合物中的聚酰胺树脂的量没有特别限制,相对于与后述的离聚物树脂的总和优选包含0.1~99.9重量%。相对于与离聚物树脂的总和,更优选为0.5~99.0重量%的范围,进一步优选为1.0~95.0重量%的范围。通过设为该范围,可得到满足作为树脂的成型性等所要求的物性且耐冲击性更优异的聚酰胺树脂组合物。
36.2.离聚物
37.本发明的离聚物的特征在于:以包含源自乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)和源自具有羧基和/或二羧酸酐基团的单体的结构单元(b)作为必要构成单元并且这些结构单元基本上直链状共聚、优选无规共聚而得到的共聚物(p)作为前体树脂,该结构单元(b)的羧基和/或二羧酸酐基团的至少一部分被转换为含有选自元素周期表第1族、2族或12族中的至少1种金属离子的含金属羧酸盐。
38.(1)结构单元(a)
39.结构单元(a)为选自由源自乙烯的结构单元及源自碳数3~20的α
‑
烯烃的结构单元组成的组中的至少1种结构单元。
40.本发明的α
‑
烯烃为由结构式:ch2=chr
18
表示的碳数3~20的α
‑
烯烃(r
18
为碳数1~18的烃基,可以为直链结构也可以具有分支)。α
‑
烯烃的碳数更优选为3~12。
41.作为结构单元(a)的具体例子,可列举出乙烯、丙烯、1
‑
丁烯、1
‑
戊烯、1
‑
己烯、1
‑
辛烯、1
‑
癸烯、3
‑
甲基
‑1‑
丁烯及4
‑
甲基
‑1‑
戊烯等,可以为乙烯。
42.另外,离聚物中所含的结构单元(a)可以是1种也可以是多种。作为2种的组合,可列举出乙烯
‑
丙烯、乙烯
‑1‑
丁烯、乙烯
‑1‑
己烯、乙烯
‑1‑
辛烯、丙烯
‑1‑
丁烯、丙烯
‑1‑
己烯及丙烯
‑1‑
辛烯等。作为3种的组合,可列举出乙烯
‑
丙烯
‑1‑
丁烯、乙烯
‑
丙烯
‑1‑
己烯、乙烯
‑
丙烯
‑1‑
辛烯、丙烯
‑1‑
丁烯
‑
己烯及丙烯
‑1‑
丁烯
‑1‑
辛烯等。
43.本发明中,作为结构单元(a)优选必须包含乙烯,根据需要可以进一步包含1种以上的碳数3~20的α
‑
烯烃。
44.相对于结构单元(a)的总mol,结构单元(a)中乙烯的量可以为65~100mol%,也可以为70~100mol%。
45.(2)结构单元(b)
46.结构单元(b)为源自具有羧基和/或二羧酸酐基团的单体的结构单元。
47.作为具有羧基的单体,具体而言,可列举出丙烯酸、甲基丙烯酸、马来酸、富马酸、四氢邻苯二甲酸、衣康酸、柠康酸、巴豆酸、异巴豆酸、降冰片烯二羧酸,双环[2.2.1]庚
‑2‑
烯
‑
5,6
‑
二羧酸等不饱和羧酸;作为具有二羧酸酐基团的单体,可列举出马来酸酐、衣康酸酐、柠康酸酐、四氢邻苯二甲酸酐、5
‑
降冰片烯
‑
2,3
‑
二羧酸酐、3,6
‑
环氧
‑
1,2,3,6
‑
四氢邻苯二甲酸酐、四环[6.2.1.1
3,6
.0
2,7
]十二
‑9‑
烯
‑
4,5
‑
二羧酸酐,2,7
‑
辛二烯
‑1‑
基琥珀酸酐等不饱和二羧酸酐。
[0048]
作为具体化合物,可列举出丙烯酸、甲基丙烯酸、5
‑
降冰片烯
‑
2,3
‑
二羧酸酐,特别是可以为丙烯酸。
[0049]
另外,具有羧基和/或二羧酸酐基团的单体可以为1种,也可以为多种。
[0050]
需要说明的是,二羧酸酐基团有时会与空气中的水分反应而开环、一部分变为二羧酸,但在不脱离本发明主旨的范围内,二羧酸酐基团也可以开环。
[0051]
(3)其它结构单元(c)
[0052]
本发明的共聚物(p)也可以包含除结构单元(a)及结构单元(b)所示结构单元以外的结构单元(c)(以下有时记作“任意单体(c)”)。提供结构单元(c)的单体,可以使用任意单体,只要不与提供结构单元(a)及结构单元(b)的单体相同即可。提供结构单元(c)的任意单体只要为分子结构中具有1个以上碳
‑
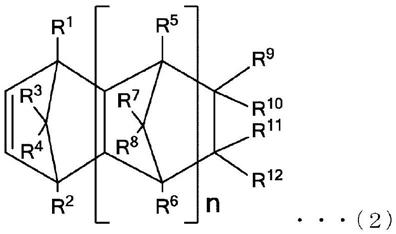
碳双键的化合物就没有限定,例如可列举出下述通式(1)所示的非环状单体、下述通式(2)所示的环状单体等。
[0053]
·
非环状单体
[0054][0055]
[通式(1)中,t1~t3各自独立地为选自由氢原子、碳数1~20的烃基、被羟基取代的碳数1~20的烃基、被碳数1~20的烷氧基取代的碳数2~20的烃基、被碳数2~20的酯基取代的碳数3~20的烃基、被卤原子取代的碳数1~20的烃基、碳数1~20的烷氧基、碳数6~20的芳基、碳数2~20的酯基、碳数3~20的甲硅烷基、卤原子或氰基组成的组中的取代基,
[0056]
t4为选自由被羟基取代的碳数1~20的烃基、被碳数1~20的烷氧基取代的碳数2~20的烃基、被碳数2~20的酯基取代的碳数3~20的烃基、被卤原子取代的碳数1~20的烃基、碳数1~20的烷氧基、碳数6~20的芳基、碳数2~20的酯基、碳数3~20的甲硅烷基、卤原子或氰基组成的组中的取代基。]
[0057]
本发明的离聚物中,t1及t2可以为氢原子,t3可以为氢原子或甲基,t4可以为碳数2~20的酯基。
[0058]
t1~t4涉及的烃基、取代烷氧基、取代酯基、烷氧基、芳基、酯基、甲硅烷基所具有的碳骨架任选具有分支、环和/或不饱和键。
[0059]
t1~t4涉及的烃基的碳数的下限值为1以上即可,上限值为20以下即可,可以为10以下。
[0060]
t1~t4涉及的取代烷氧基的碳数的下限值为1以上即可,上限值为20以下即可,可以为10以下。
[0061]
t1~t4涉及的取代酯基的碳数的下限值为2以上即可,上限值为20以下即可,可以为10以下。
[0062]
t1~t4涉及的烷氧基的碳数的下限值为1以上即可,上限值为20以下即可,可以为10以下。
[0063]
t1~t4涉及的芳基的碳数的下限值为6以上即可,上限值为20以下即可,可以为11以下。
[0064]
t1~t4涉及的酯基的碳数的下限值为2以上即可,上限值为20以下即可,可以为10以下。
[0065]
t1~t4涉及的甲硅烷基的碳数的下限值为3以上即可,上限值为18以下即可,可以为12以下。作为甲硅烷基,可列举出三甲基甲硅烷基、三乙基甲硅烷基、三正丙基甲硅烷基、三异丙基甲硅烷基、二甲基苯基甲硅烷基、甲基二苯基甲硅烷基及三苯基甲硅烷基等。
[0066]
作为非环状单体,具体而言,可列举出(甲基)丙烯酸酯等。
[0067]
本发明的(甲基)丙烯酸酯为结构式:ch2=c(r
21
)co2(r
22
)所示的化合物。其中,r
21
为氢原子或碳数1~10的烃基,任选具有分支、环和/或不饱和键。r
22
为碳数1~20的烃基,任选具有分支、环和/或不饱和键。并且,在r
22
内的任意位置任选具有杂原子。
[0068]
作为(甲基)丙烯酸酯,可列举出r
21
为氢原子或碳数1~5的烃基的(甲基)丙烯酸酯。另外,可列举出r
21
为氢原子的丙烯酸酯或r
21
为甲基的甲基丙烯酸酯。
[0069]
作为(甲基)丙烯酸酯的具体例子,例如可列举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸正丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸正丁酯、(甲基)丙烯酸异丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸戊酯、(甲基)丙烯酸己酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸辛酯、(甲基)丙烯酸2
‑
乙基己酯、(甲基)丙烯酸壬酯、(甲基)丙烯酸癸酯、(甲基)丙烯酸十二烷基酯、(甲基)丙烯酸十八烷基酯、(甲基)丙烯酸苯酯、(甲基)丙烯酸甲苯酯和(甲基)丙烯酸苄酯等。
[0070]
作为具体化合物,可列举出丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丁酯(nba)、丙烯酸异丁酯(iba)、丙烯酸叔丁酯(tba)及丙烯酸2
‑
乙基己酯等,特别是可以为丙烯酸正丁酯(nba)、丙烯酸异丁酯(iba)及丙烯酸叔丁酯(tba)。
[0071]
需要说明的是,非环状单体可以使用1种,也可以使用多种。
[0072]
·
环状单体
[0073][0074]
[通式(2)中,r1~r
12
可以彼此相同或不同,选自由氢原子、卤原子及碳数1~20的烃基组成的组,r9和r
10
以及r
11
和r
12
分别任选一体化形成2价的有机基团,r9或r
10
与r
11
或r
12
任选彼此形成环。
[0075]
另外,n表示0或正整数,n为2以上时,r5~r8在各自的重复单元、可以彼此相同或不同。]
[0076]
作为环状单体,可列举出降冰片烯系烯烃等,可列举出降冰片烯、降冰片烯、乙烯基降冰片烯、乙叉基降冰片烯、降冰片二烯、四环十二碳烯、三环[4.3.0.1
2,5
]十
‑1‑
烯、三环[4.3.0.1
2,5
]十
‑3‑
烯等具有环状烯烃骨架的化合物等,可以为2
‑
降冰片烯(nb)及四环[6.2.1.1
3,6
.0
2,7
]十二
‑4‑
烯等。
[0077]
(4)金属离子
[0078]
离聚物中含有与上述(2)的结构单元(b)所具有的羧基和/或二羧酸酐基团形成盐的金属离子。作为羧酸盐基的金属离子,可列举出选自由元素周期表第1族、第2族及第12族组成的组中的族的一价或二价金属离子。作为金属离子,具体而言,可列举出锂(li)、钠(na)、钾(k)、铷(rb)、镁(mg)、钙(ca)及锌(zn)的离子等,从操作难易度的观点出发,特别是可以为钠(na)或锌(zn)的离子。
[0079]
羧酸盐基例如可通过将共聚物的酯基水解或加热分解后或者在使其水解或加热
分解的同时与含有元素周期表第1族、2族或12族的金属离子的化合物反应,使共聚物中的酯基部分转换为含金属羧酸盐来得到。
[0080]
需要说明的是,金属离子可以为1种,也可以为多种。
[0081]
(5)共聚物(p)
[0082]
作为本发明中使用的离聚物的前体树脂的共聚物(p)的特征在于:包含源自乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)和源自具有羧基和/或二羧酸酐基团的单体的结构单元(b)作为必要构成单元,并且这些结构单元基本上直链状共聚、优选无规共聚。“基本上直链状”是指,共聚物不具有分支或分支结构出现的频率小、可将共聚物视为直链状的状态。具体而言,是指在下述条件下测定的共聚物的相位角δ为50度以上的状态。
[0083]
本发明的共聚物需要包含结构单元(a)和结构单元(b)各含1种以上、共2种以上的单体单元,也可以包含其它结构单元(c)。
[0084]
对本发明的共聚物的结构单元与结构单元量进行说明。
[0085]
将源自各1分子的乙烯和/或碳数3~20的α
‑
烯烃(a)、具有羧基和/或二羧酸酐基团的单体(b)及任意单体(c)的结构定义为共聚物中的1结构单元。
[0086]
并且,结构单元量是将共聚物中的结构单元整体设为100mol%时以mol%表示的各结构单元的比率。
[0087]
乙烯和/或碳数3~20的α
‑
烯烃(a)的结构单元量:
[0088]
本发明的结构单元(a)的结构单元量的下限为60.000mol%以上,优选为70.000mol%以上,更优选为80.000mol%以上,进一步优选为85.000mol%以上,更进一步优选为90.000mol%以上,特别优选为91.200mol%以上;上限选自97.999mol%以下、优选97.990mol%以下、更优选97.980mol%以下、进一步优选96.980mol%以下、更进一步优选96.900mol%以下、特别优选92.700mol%以下。
[0089]
若源自乙烯和/或碳数3~20的α
‑
烯烃(a)的结构单元量少于60.000mol%,则聚酰胺树脂组合物的刚性变差,多于97.999mol%,则存在聚酰胺树脂组合物的改善效果降低的情况。
[0090]
·
源自具有羧基和/或二羧酸酐基团的单体(b)的结构单元量:
[0091]
本发明的结构单元(b)的结构单元量的下限为2.0mol%以上,优选为2.9mol%以上,更优选为5.2mol%以上;上限选自20.0mol%以下、优选15.0mol%以下、更优选10.0mol%以下、进一步优选8.0mol%以下、特别优选5.4mol%以下。
[0092]
若源自具有羧基和/或二羧酸酐基团的单体(b)的结构单元量少于2.0mol%,则以共聚物(p)为前体树脂制造的离聚物相对于聚酰胺树脂的分散性不充分,损害耐冲击性的改善效果,若多于20.0mol%,则存在无法得到聚酰胺树脂组合物的充分的机械物性的情况。
[0093]
·
任意单体(c)的结构单元量:
[0094]
对于本发明的结构单元(c)的结构单元量,包含结构单元(c)的情况下,下限为0.001mol%以上,优选为0.010mol%以上,更优选为0.020mol%以上,进一步优选为0.100mol%以上,更进一步优选为1.000mol%以上,特别优选为1.900mol%以上;上限选自20.000mol%以下、优选15.000mol%以下、更优选10.000mol%以下、进一步优选5.000mol%以下、特别优选3.600mol%以下。
[0095]
并且,使用的任意单体可以单独使用,也可以2种以上组合使用。
[0096]
·
共聚物的每1000个碳的分支数:
[0097]
聚烯烃的分子结构中,甲基、乙基和丁基等碳数1~4左右的短碳链会以分支的形式出现。本发明的共聚物具有不具备分支或分支结构出现的频率小、可视为直链状的结构。
[0098]
共聚物中,从提高弹性模量、得到充分的机械物性的观点出发,每1000个碳,由
13
c
‑
nmr算出的甲基分支数的上限可以为50个以下,也可以为5.0个以下,也可以为1.0个以下,也可以为0.5个以下。甲基分支数的下限没有特别限定,越少越好。另外,每1000个碳,乙基分支数的上限可以为3.0个以下,也可以为2.0个以下,也可以为1.0个以下,也可以为0.5个以下。乙基分支数的下限没有特别限定,越少越好。并且,每1000个碳,丁基分支数的上限可以为7.0个以下,也可以为5.0个以下,也可以为3.0个以下,也可以为0.5个以下。丁基分支数的下限没有特别限定,越少越好。
[0099]
共聚物中的源自羧基和/或二羧酸酐基团单体的结构单元及分支数的测定方法:
[0100]
本发明的多元共聚物中的源自羧基和/或二羧酸酐基团单体的结构单元、每1000个碳的分支数使用
13
c
‑
nmr谱来求出。
13
c
‑
nmr通过以下方法进行测定。
[0101]
将试样200~300mg与邻二氯苯(c6h4cl2)和氘代溴苯(c6d5br)的混合溶剂(c6h4cl2/c6d5br=2/1(体积比))2.4ml以及作为化学位移标准物质的六甲基二硅氧烷一起放入内径10mmφ的nmr试样管中,进行氮置换后封管,进行加热溶解制成均匀的溶液作为nmr测定试样。
[0102]
nmr测定使用装有10mmφ冷冻探针的brukerjapan株式会社的av400m型nmr装置在120℃下进行。
[0103]
13
c
‑
nmr在试样温度120℃、脉冲角90
°
、脉冲间隔51.5秒、累计次数512次以上的条件下使用反转门控去耦法进行测定。
[0104]
化学位移是将六甲基二硅氧烷的
13
c信号设定为1.98ppm,其它基于
13
c的信号的化学位移以其为基准。
[0105]
得到的
13
c
‑
nmr中,可以通过识别共聚物具有的单体或分支所特有的信号,比较其强度来分析共聚物中的各单体的结构单元量及分支数。单体或分支特有的信号的位置可以参考公知的资料,或者也可以根据试样自行识别。该分析方法是本领域技术人员通常可以进行的。
[0106]
·
重均分子量(mw)与分子量分布(mw/mn):
[0107]
本发明的共聚物的重均分子量(mw)的下限通常为1,000以上,优选为6,000以上,上限通常为2,000,000以下,优选为1,500,000以下,进一步优选为1,000,000以下,特别优选为800,000以下,最优选为56,000以下。
[0108]
若mw小于1000,则共聚物的机械的强度、耐冲击性等物性不充分,若mw超过2,000,000,则存在共聚物的熔融粘度变得非常高,难以进行共聚物的成型加工的情况。
[0109]
本发明的共聚物的重均分子量(mw)与数均分子量(mn)的比(mw/mn)通常为1.5~4.0,优选为1.6~3.5,进一步优选为1.9~2.3的范围。若mw/mn小于1.5,则包括共聚物的成型在内的各加工性不充分,若超过4.0,则存在共聚物的机械物性变差的情况。
[0110]
另外,本发明中,有时将(mw/mn)称作分子量分布参数。
[0111]
本发明的重均分子量(mw)及数均分子量(mn)可通过凝胶渗透色谱法(gpc)求出。
另外,对于分子量分布参数(mw/mn),通过凝胶渗透色谱法(gpc)进一步求出数均分子量(mn),算出mw与mn的比、mw/mn。
[0112]
本发明的gpc的测定方法的1个例子如下。
[0113]
(测定条件)
[0114]
使用机型:waters公司制150c
[0115]
检测器:foxboro公司制miran1a
·
ir检测器(测定波长:3.42μm)
[0116]
测定温度:140℃
[0117]
溶剂:邻二氯苯(odcb)
[0118]
柱:昭和电工株式会社制ad806m/s(3根)
[0119]
流速:1.0ml/分钟
[0120]
注入量:0.2ml
[0121]
(试样的制备)
[0122]
对于试样,使用odcb(含0.5mg/ml的bht(2,6
‑
二叔丁基
‑4‑
甲基苯酚))制备1mg/ml的溶液,在140℃下用约1小时使其溶解。
[0123]
(分子量(m)的计算)
[0124]
通过标准聚苯乙烯法来进行,使用预先制作的基于标准聚苯乙烯的校准曲线来进行由保持容量至分子量的换算。使用的标准聚苯乙烯均为东曹公司制的商品名f380、f288、f128、f80、f40、f20、f10、f4、f1、a5000、a2500、a1000的产品。注入各标准聚苯乙烯以为0.5mg/ml的方式溶解于odcb(含0.5mg/mlbht)而得到的溶液0.2ml,制作校准曲线。校准曲线使用利用最小二乘法近似得到的三次式。换算为分子量(m)时使用的粘度式[η]=k
×
mα使用以下数值。
[0125]
聚苯乙烯(ps):k=1.38
×
10
‑4,α=0.7
[0126]
聚乙烯(pe):k=3.92
×
10
‑4,α=0.733
[0127]
聚丙烯(pp):k=1.03
×
10
‑4,α=0.78
[0128]
·
熔点(tm、℃):
[0129]
本发明的共聚物的熔点由通过差示扫描量热计(dsc)测定的吸热曲线的最大峰温度来表示。对于最大峰温度,在dsc测量中,在纵轴为热流(mw)、横轴为温度(℃)时得到的吸热曲线中出现多个峰的情况下,是指距基线的高度最大的峰的温度,在峰为1个的情况下,是指该峰的温度。
[0130]
熔点优选为50℃~140℃,进一步优选为60℃~138℃,最优选为70℃~135℃。若低于该范围,则耐热性不充分,若高于该范围,则存在聚酰胺树脂组合物的刚性变差的情况。
[0131]
共聚物的熔点例如可以通过使用sii nanotechnology inc.制的dsc(dsc7020),将约5.0mg试样装入铝盘中,以10℃/分钟升温至200℃,在200℃下等温保持5分钟后,以10℃/分钟降温至20℃,在20℃下等温保持5分钟后,再次以10℃/分钟升温至200℃时的吸收曲线来求出。
[0132]
·
结晶度(%):
[0133]
本发明的共聚物中,通过差示扫描量热测定(dsc)观测到的结晶度没有特别限定,但优选超过0%。更优选超过5%,进一步优选为7%以上。结晶度为0%时,存在共聚物的韧
性变得不充分的情况。另外,结晶度为透明性的指标,共聚物的结晶度越低,可以判断其透明性越优异。在要求透明性的用途中,共聚物的结晶度优选为30%以下,进一步优选为25%以下,特别优选为25%以下,最优选为24%以下。
[0134]
共聚物的结晶度例如可以如下获得:由通过与上述熔点测定相同步骤的dsc测定得到的熔解吸热峰面积求出熔解热(δh),将其熔解热除以高密度聚乙烯(hdpe)的完全结晶的熔解热293j/g,由此求出结晶度。
[0135]
·
共聚物的分子结构:
[0136]
本发明的共聚物的分子链末端可以为乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a),也可以为具有羧基和/或二羧酸酐基团的单体的结构单元(b),也可以为任意单体的结构单元(c)。
[0137]
另外,本发明的共聚物可列举出乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)、具有羧基和/或二羧酸酐基团的单体的结构单元(b)及任意单体的结构单元(c)的无规共聚物、嵌段共聚物及接枝共聚物等。其中,可以为可含有大量结构单元(b)的无规共聚物。
[0138]
通常的三元系共聚物的分子结构例(1)如下所示。
[0139]
无规共聚物是指如下所示的分子结构例(1)中的乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)、具有羧基和/或二羧酸酐基团的单体的结构单元(b)和任意单体的结构单元(c)在某任意分子链中的位置中各结构单元出现的概率与其相邻的结构单元的种类无关的共聚物。
[0140]
如下所述,共聚物的分子结构例(1)中,乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)、具有羧基和/或二羧酸酐基团的单体的结构单元(b)和任意单体的结构单元(c)形成无规共聚物。
[0141]
‑
abgaaabbcbaabaccaa
‑
[0142]
···
分子结构例(1)
[0143]
需要说明的是,作为参考也示出了通过接枝改性引入了具有羧基和/或二羧酸酐基团的单体的结构单元(b)的共聚物的分子结构例(2),其中,乙烯和/或碳数3~20的α
‑
烯烃的结构单元(a)和任意单体的结构单元(c)共聚而得到的共聚物的一部分被具有羧基和/或二羧酸酐基团的单体的结构单元(b)接枝改性。
[0144][0145]
另外,共聚物中的无规共聚性可通过各种方法进行确认,但由共聚物的共聚单体含量与熔点的关系判别无规共聚性的方法在“日本特开2015
‑
163691号公报”及“日本特开2016
‑
079408”中有详细说明。根据上述文献,共聚物的熔点(tm、℃)高于
‑
3.74
×
[z] 130(其中,[z]为共聚单体含量/mol%)时,可判断无规性低。
[0146]
作为无规共聚物的本发明的共聚物中,优选通过差示扫描量热测定(dsc)观测到的熔点(tm、℃)与具有羧基和/或二羧酸酐基团的单体的结构单元(b)及任意单体的结构单元(c)的总含量[z](mol%)满足下述式(i)。
[0147]
50<tm<
‑
3.74
×
[z] 130
···
(i)
[0148]
共聚物的熔点(tm,℃)高于
‑
3.74
×
[z] 130(℃)时,无规共聚性低,故冲击强度等机械物性差,熔点低于50℃时,存在耐热性差的情况。
[0149]
并且,本发明的共聚物从使其分子结构为直链状的观点出发,优选是在过渡金属催化剂的存在下制造的。
[0150]
需要说明的是,已知共聚物的分子结构根据制造方法而不同,例如基于高压自由基聚合法工艺的聚合、使用金属催化剂的聚合等。
[0151]
该分子结构的差异可通过选择制造方法来控制,例如如日本特开2010
‑
150532号公报中记载的那样,也可以通过用旋转式流变仪测定的复数模量来推定其分子结构。
[0152]
·
复数模量的绝对值g*=0.1mpa下的相位角δ:
[0153]
本发明的共聚物的特征在于用旋转式流变仪测定的复数模量的绝对值g*=0.1mpa下的相位角δ为50~75度。相位角δ的下限可以为51度以上,也可以为54度以上,也可以为56度以上,也可以为58度以上。相位角δ的上限可以为75度以下,也可以为70度以下。
[0154]
更具体而言,用旋转式流变仪测定的复数模量的绝对值g*=0.1mpa下的相位角δ(g*=0.1mpa)为50度以上时,共聚物的分子结构显示其为呈直链状结构且完全不包含长支链的结构、或者在不影响机械强度的范围内包含少量长支链的基本上为直链状的结构。
[0155]
另外,用旋转式流变仪测定的复数模量的绝对值g*=0.1mpa下的相位角δ(g*=0.1mpa)低于50度时,共聚物的分子结构显示过多包含长支链的结构,机械强度变差。
[0156]
用旋转式流变仪测定的复数模量的绝对值g*=0.1mpa下的相位角δ受分子量分布和长支链这两者的影响。但是,只有在mw/mn≤4、更优选mw/mn≤3的共聚物中,为长支链的量的指标,其分子结构中所含的长支链越多,δ(g*=0.1mpa)值越小。需要说明的是,只要共聚物的mw/mn为1.5以上,即使该分子结构为不含长支链的结构的情况下,δ(g*=0.1mpa)值也不会超过75度。
[0157]
复数模量的测定方法如下。
[0158]
将试样置于厚度1.0mm的热压用模具中,在表面温度180℃的热压机中预热5分钟后,重复进行加压和减压,使熔融树脂中的残留气体脱气,进一步地以4.9mpa进行加压,保持5分钟。然后,将试样移至表面温度25℃的压制机中,在4.9mpa的压力下保持3分钟来使其冷却,制成厚度为约1.0mm的压制板。将压制板加工为直径25mm圆形,将其作为样品,作为动态粘弹性特性的测定装置,使用rheometrics公司制ares型旋转式流变仪,在氮气气氛下采用以下条件测定动态粘弹性。
[0159]
·
板:φ25mm平行板
[0160]
·
温度:160℃
[0161]
·
应变量:10%
[0162]
·
测定角频率范围:1.0
×
10
‑2~1.0
×
102rad/s
[0163]
·
测定间隔:5点/decade
[0164]
相对于复数模量的绝对值g*(pa)的常用对数logg*绘制相位角δ,将相当于logg*=5.0的点的δ(度)的值设为δ(g*=0.1mpa)。当测定点中没有相当于logg*=5.0的点时,使用logg*=5.0前后的2个点,利用线性插值求出logg*=5.0的δ值。另外,测定点均为logg*<5时,从logg*值大的一侧取3个点的值,利用2次曲线外推求出logg*=5.0的δ值。
[0165]
·
关于共聚物的制造
[0166]
本发明的共聚物从使其分子结构为直链状的观点出发,优选是在过渡金属催化剂的存在下制造的。
[0167]
·
聚合催化剂
[0168]
对于本发明的共聚物的制造中使用的聚合催化剂的种类,只要为能够使结构单元(a)、结构单元(b)及任意结构单元(c)共聚的催化剂就没有特别限定,例如可列举出具有螯合性配体的第5~11族的过渡金属化合物。
[0169]
作为优选的过渡金属的具体例子,可列举出钒原子、铌原子、钽原子、铬原子、钼原子、钨原子、锰原子、铁原子、铂原子、钌原子、钴原子、铑原子、镍原子、钯原子、铜原子等。其中,优选为第8~11族的过渡金属,更优选为第10族的过渡金属,特别优选为镍(ni)、钯(pd)。这些金属可以使用1种,也可以同时使用多种。
[0170]
螯合性配体具有选自由p、n、o及s组成的组中的至少2个原子,包含为二齿配位(bidentate)或多齿配位(multidentate)的配体,为电中性或阴离子性。brookhart等的综述中,列举了螯合性配体的结构(chem.rev.,2000,100,1169)。
[0171]
作为螯合性配体,优选地可列举出二齿阴离子性p、o配体。作为二齿阴离子性p、o配体,例如可列举出磷磺酸、磷羧酸、磷酚、磷烯醇化物。除此以外,作为螯合性配体,可列举出二齿阴离子性n、o配体。作为二齿阴离子性n、o配体,例如可列举出水杨醛亚胺、吡啶羧酸。除此以外,作为螯合性配体,可列举出二亚胺配体、二苯氧化物配体及二酰胺配体等。
[0172]
由螯合性配体得到的金属络合物的结构由配位有任选具有取代基的芳基膦化合物、芳基胂化合物或芳基锑化合物的下述结构式(c1)或(c2)表示。
[0173][0174]
[结构式(c1)及结构式(c2)中,
[0175]
m表示属于元素周期表第5~11族中任一者的过渡金属,即前述的各种过渡金属。
[0176]
x1表示氧、硫、
‑
so3‑
或
‑
co2‑
。
[0177]
y1表示碳或硅。
[0178]
n表示0或1的整数。
[0179]
e1表示磷、砷或锑。
[0180]
r
53
和r
54
各自独立地表示氢或碳数1~30的任选含有杂原子的烃基。
[0181]
r
55
各自独立地表示氢、卤素或碳数1~30的任选含有杂原子的烃基。
[0182]
r
56
及r
57
各自独立地表示氢、卤素、碳数1~30的任选含有杂原子的烃基、or
52
、co2r
52
、co2m’、c(o)n(r
51
)2、c(o)r
52
、sr
52
、so2r
52
、sor
52
、oso2r
52
、p(o)(or
52
)2‑
y
(r
51
)
y
、cn、nhr
52
、n(r
52
)2、si(or
51
)3‑
x
(r
51
)
x
、osi(or
51
)3‑
x
(r
51
)
x
、no2、so3m’、po3m
’2、p(o)(or
52
)2m’或含环氧基的基团。
[0183]
r
51
表示氢或碳数1~20的烃基。
[0184]
r
52
表示碳数1~20的烃基。
[0185]
m’表示碱金属、碱土金属、铵、季铵或鏻,x表示0~3的整数,y表示0~2的整数。
[0186]
需要说明的是,r
56
和r
57
任选相互连接,形成脂环式环、芳香族环或者含有选自氧、氮或硫的杂原子的杂环。此时,环元数为5~8,该环上可具有取代基,也可以不具有取代基。
[0187]
l1表示与m配位的配体。
[0188]
另外,r
53
和l1任选彼此键合形成环。]
[0189]
更优选地,作为聚合催化剂的络合物为下述结构式(c3)所示的过渡金属络合物。
[0190][0191]
[结构式(c3)中,
[0192]
m表示属于元素周期表第5~11族中任一者的过渡金属,即前述的各种过渡金属。
[0193]
x1表示氧、硫、
‑
so3‑
或
‑
co2‑
。
[0194]
y1表示碳或硅。
[0195]
n表示0或1的整数。
[0196]
e1表示磷、砷或锑。
[0197]
r
53
和r
54
各自独立地表示氢或碳数1~30的任选含有杂原子的烃基。
[0198]
r
55
各自独立地表示氢、卤素或碳数1~30的任选含有杂原子的烃基。
[0199]
r
58
、r
59
、r
60
及r
61
各自独立地表示氢、卤素、碳数1~30的任选含有杂原子的烃基、or
52
、co2r
52
、co2m’、c(o)n(r
51
)2、c(o)r
52
、sr
52
、so2r
52
、sor
52
、oso2r
52
、p(o)(or
52
)2‑
y
(r
51
)
y
、cn、nhr
52
、n(r
52
)2、si(or
51
)3‑
x
(r
51
)
x
、osi(or
51
)3‑
x
(r
51
)
x
、no2、so3m’、po3m
’2、p(o)(or
52
)2m’或含有环氧基的基团。
[0200]
r
51
表示氢或碳数1~20的烃基。
[0201]
r
52
表示碳数1~20的烃基。
[0202]
m’表示碱金属、碱土金属、铵、季铵或鏻,x表示0~3的整数,y表示0~2的整数。
[0203]
需要说明的是,适当地选自r
58
~r
61
的多个基团任选相互连接,形成脂环式环、芳香族环或者含有选自氧、氮或硫的杂原子的杂环。此时,环元数为5~8,该环上可具有取代基,也可以不具有取代基。
[0204]
l1表示与m配位的配体。
[0205]
另外,r
53
和l1任选彼此键合形成环。]
[0206]
此处,作为具有螯合性配体的第5~11族的过渡金属化合物的催化剂,代表性地,已知有所谓的shop系催化剂、drent系催化剂等催化剂。
[0207]
shop系催化剂为具有任选具有取代基的芳基的磷系配体配位于镍金属而得到的催化剂(例如参见wo2010
‑
050256号公报)。
[0208]
另外,drent系催化剂为具有任选具有取代基的芳基的磷系配体配位于钯金属而得到的催化剂(例如参见日本特开2010
‑
202647号公报)。
[0209]
·
共聚物的聚合方法:
[0210]
本发明的共聚物的聚合方法没有限定。
[0211]
作为聚合方法,可列举出至少一部分生成聚合物在介质中变为浆料的浆料聚合、将液化的单体自身作为介质的本体聚合、在气化的单体中进行的气相聚合或使生成聚合物的至少一部分溶解于高温高压下液化的单体中的高压离子聚合等。
[0212]
作为聚合形式,可以为间歇聚合、半间歇聚合或连续聚合中的任一形式。
[0213]
另外,也可以进行活性聚合,或者也可以在引起链转移的同时进行聚合。
[0214]
并且,聚合时,可以同时使用所谓的链穿梭剂(chain shuttling agent、csa),进行链穿梭(chain shuttling)反应、配位链转移聚合(coordinative chain transfer polymerization、cctp)。
[0215]
具体的制造工艺及条件例如在日本特开2010
‑
260913号公报及日本特开2010
‑
202647号公报等中有公开。
[0216]
·
在共聚物中引入羧基和/或二羧酸酐基团的方法:
[0217]
在本发明的共聚物中引入羧基和/或二羧酸酐基团的方法没有特别限定。
[0218]
在不脱离本发明的主旨的范围内,可以通过各种方法引入羧基和/或二羧酸酐基团。
[0219]
羧基和/或二羧酸酐基团的引入方法例如可列举出:直接使具有羧基和/或二羧酸酐基团的共聚单体共聚的方法;使其它单体共聚后,通过改性引入羧基和/或二羧酸酐基团的方法等。
[0220]
作为通过改性引入羧基和/或二羧酸酐基团的方法,例如引入羧酸的情况下,可列举出使丙烯酸酯共聚后,进行水解使其变为羧酸的方法;或使丙烯酸叔丁酯共聚后,通过加热分解使其变为羧酸的方法等。
[0221]
作为上述水解或加热分解时促进反应的添加剂,可以使用以往公知的酸/碱催化剂。作为酸/碱催化剂,没有特别限制,例如可以适当使用氢氧化钠、氢氧化钾、氢氧化锂等碱金属或碱土金属的氢氧化物;碳酸氢钠、碳酸钠等碱金属、碱土金属的碳酸盐;蒙脱石等
固体酸;盐酸、硝酸、硫酸等无机酸;甲酸、乙酸、苯甲酸、柠檬酸、对甲苯磺酸、三氟乙酸、三氟甲磺酸等有机酸等。从反应促进效果、价格、装置腐蚀性等观点出发,优选氢氧化钠、氢氧化钾、碳酸钠、对甲苯磺酸、三氟乙酸,更优选对甲苯磺酸、三氟乙酸。
[0222]
(6)离聚物
[0223]
本发明的离聚物为本发明的共聚物的结构单元(b)的羧基和/或二羧酸酐基团的至少一部分通过前述金属离子被转换为含有选自元素周期表第1族、2族或12族中的至少1种金属离子的含金属羧酸盐的、基本上具有直链状结构的离聚物。
[0224]
·
离聚物的结构
[0225]
本发明的离聚物与上述本发明的共聚物同样地为基本上具有直链状结构的无规共聚物,因此结构相关的参数优选为与上述共聚物的参数相同的范围。即,对于用旋转式流变仪测定的复数模量的绝对值g*=0.1mpa下的相位角δ、熔点(tm,℃)、结晶度(%),使用与上述共聚物相同的优选方案。如后文中所述,离聚物是使金属盐作用于前体树脂而得到的,此时通常不会发生切断聚合物的分子链的反应。因此,共聚单体的摩尔比、分支的程度、无规性等结构相关的参数通常在前体树脂与离聚物之间是得以保存的。
[0226]
·
中和度(mol%)
[0227]
作为离聚物中金属离子的含量,优选含有使作为前体树脂的共聚物中的羧基和/或二羧酸酐基团的至少一部分或全部中和的量,优选的中和度(平均中和度)为5~95mol%,更优选为10~90mol%,进一步优选为20~80mol%。
[0228]
中和度高时,虽然离聚物的拉伸强度及拉伸断裂应力高,拉伸断裂应变小,但存在离聚物的熔体流动速率(mfr)降低的倾向。另一方面,中和度低时,虽然可得到适度的mfr的离聚物,在成型性方面变得更好,但存在聚酰胺树脂组合物的耐冲击性改善效果不充分的情况。
[0229]
需要说明的是,中和度可根据羧基和/或二羧酸酐基团的量与加入的金属离子的摩尔比来计算。
[0230]
·
离聚物的制造方法
[0231]
本发明的离聚物可通过经过如下转换工序来得到:利用含有选自元素周期表第1族、2族或12族中的至少1种金属离子的金属盐对由如上所述的在共聚物中引入羧基和/或二羧酸酐基团的方法得到的乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸的共聚物进行处理,使其转换为含金属羧酸盐。另外,本发明的离聚物也可通过经过如下加热转换工序来得到:对乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸酯共聚物进行加热,将该共聚物中的至少一部分酯基转换为含有选自元素周期表第1族、2族或12族中的至少1种金属离子的含金属羧酸盐。
[0232]
在聚合物中引入羧基和/或二羧酸酐基团后制造离聚物的情况下,该制造方法例如如下所述。即,根据情况对捕捉乙烯/甲基丙烯酸(maa)共聚物等的金属离子的物质和金属盐进行加热和混炼,由此制造金属离子供给源,接着在离聚物的前体树脂中加入形成期望的中和度的量的该金属离子供给源,进行混炼,由此得到离聚物。
[0233]
另外,加热转换工序中,(i)可以在对乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸酯共聚物进行加热,通过水解或加热分解制成乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸共聚物后,使其与含有元素周期表第1族、第2族或第12族的金属离子的化合物反应,由此将
该乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸共聚物中的羧酸转换为该含金属羧酸盐。或者(ii)也可以对乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸酯共聚物进行加热,一边使该共聚物的酯基水解或加热分解,一边使其与含有元素周期表第1族、第2族或第12族的金属离子的化合物反应,由此将上述乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸酯共聚物中的酯基部分转换为上述含金属羧酸盐。
[0234]
并且,含有金属离子的化合物可以为元素周期表第1族、第2族或第12族的金属的氧化物、氢氧化物、碳酸盐、碳酸氢盐、乙酸盐、甲酸盐等。
[0235]
含有金属离子的化合物可以粒状或细粉状供于反应体系,也可以在水、有机溶剂中溶解或分散后供于反应体系,也可以制作以乙烯/不饱和羧酸共聚物、烯烃共聚物为基础聚合物的母料并供于反应体系。为了使反应顺利进行,优选制作母料并供于反应体系的方法。
[0236]
另外,与含有金属离子的化合物之间的反应可以通过利用排气式挤出机、班伯里密炼机、辊磨机等各种类型的装置进行熔融混炼来进行,反应可以为间歇式也可以为连续法。通过用脱气装置排出反应副产的水及二氧化碳,可使反应顺利进行,因此优选使用排气式挤出机那样的带有脱气装置的挤出机连续地进行。
[0237]
与含有金属离子的化合物反应时,为了促进反应,可注入少量的水。
[0238]
对于加热乙烯和/或碳数3~20的α
‑
烯烃/不饱和羧酸酯共聚物的温度,只要是酯变为羧酸的温度即可,加热温度过低时,酯无法转换为羧酸,过高时会进行脱羰化或共聚物的分解。因此,本发明的加热温度优选在80℃~350℃、更优选100℃~340℃、进一步优选150℃~330℃、更进一步优选200℃~320℃的范围内进行。
[0239]
反应时间根据加热温度、酯基部分的反应性等而变化,通常为1分钟~50小时,更优选为2分钟~30小时,进一步优选为2分钟~10小时,更进一步优选为2分钟~3小时,特别优选为3分钟~2小时。
[0240]
上述工序中,反应气氛没有特别限制,通常优选在非活性气体气流下进行。作为非活性气体的例子,可以使用氮、氩、二氧化碳气氛,需要说明的是,也可以存在少量的氧气或空气的混入。
[0241]
作为上述工序中使用的反应器,没有特别限制,只要为能够基本上均匀搅拌共聚物的方法就没有任何限定,可以使用装有搅拌机的玻璃容器或高压釜(ac),也可以使用布拉本德塑性计、单螺杆或双螺杆挤出机、强力螺杆式混炼机、班伯里密炼机、捏合机和辊等以往已知的任意混炼机。
[0242]
前体树脂中是否引入有金属离子并形成了离聚物可以通过测定所得树脂的ir谱分析源自羧酸(二聚体)的羰基的峰的减少来进行确认。中和度也同样地,除了前述的由摩尔比进行计算以外,可通过分析源自羧酸(二聚体)的羰基的峰的减少与源自羧酸盐基的羰基的峰的增加来进行确认。
[0243]
对于聚酰胺树脂组合物中的离聚物的量,相对于与前述聚酰胺树脂的总和,优选包含0.1~99.9重量%。更优选相对于与聚酰胺树脂的总和为0.5~99.0重量%的范围,进一步优选为1.0~95.0重量%的范围。通过设为该范围,可进一步改善聚酰胺树脂组合物的耐冲击性。
[0244]
3.聚酰胺树脂组合物
[0245]
本发明的聚酰胺树脂组合物为包含上述聚酰胺树脂(i)及离聚物树脂(ii)的组合物。本发明的聚酰胺树脂组合物可由上述2种成分构成,但在不脱离本发明主旨的范围内,为了引入其它物性,可以配混除上述聚酰胺树脂(i)及离聚物树脂(ii)以外的树脂。作为该树脂,例如可列举出高分子量聚乙烯、乙烯单独的均聚物,乙烯/α
‑
烯烃共聚物、聚丙烯等聚烯烃树脂。对于这些树脂的配混量,只要在不损害本发明的聚酰胺树脂组合物所起到的耐冲击性的范围内就没有特别限制,但优选相对于上述聚酰胺树脂(i)及离聚物树脂(ii)的总和100重量份为0~200重量份的范围。
[0246]
另外,本发明的树脂组合物在不脱离本发明的主旨的范围内,也可以配混以往公知的抗氧化剂、紫外线吸收剂、润滑剂、抗静电剂、着色剂、颜料、交联剂、发泡剂、成核剂、导电材料及填充材料等添加剂。对于这些添加剂的配混量,只要是本领域技术人员即可分别使用合适的量。
[0247]
本发明的聚酰胺树脂组合物可以通过以任意顺序将上述各成分以前述配比配混,使用单螺杆挤出机、双螺杆挤出机、超级混合机、亨舍尔混合机、班伯里密炼机、辊式密炼机、布拉本德塑性计、捏合机等通常的混炼机进行混炼、造粒来制造。此时,优选选择能够使各成分良好地分散的混炼、造粒方法,从经济性等方面出发,特别优选使用双螺杆挤出机进行混炼、造粒。混炼机可以使用基于不同混炼方法的多台机器形成多段混炼。
[0248]
4.成型品
[0249]
本发明的一个方案为使用上述聚酰胺树脂组合物的成型品。本发明的聚酰胺树脂组合物具备高耐冲击性,因此在需要物理强度的包括扰流板、进气管、进气歧管、谐振器、燃料箱、燃料加注管、燃料输送管、其它各种软管/管/罐类等汽车部件、电动工具外壳、管类等机械部件在内的电气/电子部件、家庭/办公用品、建材相关部件、家具用部件等各种用途中是有效的。另外,为了充分利用离聚物具有的特性,可以有效地用于食品用膜等包装材料、高尔夫球那样的以施加冲击为前提的产品。
[0250]
实施例
[0251]
下面列举实施例及比较例对本发明进行更具体的说明,但本发明不受这些实施例的限定。需要说明的是,实施例及比较例中的物性的测定与评价通过以下所示方法来实施。另外,表中的no data意为未测定,not detected意为低于检测极限。
[0252]
(1)熔体流动速率(mfr)
[0253]
mfr按照jis k
‑
7210(1999年)的表1
‑
条件7,在温度190℃、载荷21.18n的条件下进行测定。
[0254]
(2)拉伸冲击强度
[0255]
1)拉伸冲击强度试验样品的制作方法
[0256]
将试样置于厚度1.0mm的热压用模具中,在表面温度230℃的热压机中预热5分钟后,重复进行加压和减压,使试样熔融的同时使试样中的残留气体脱气,进一步地以4.9mpa进行加压,保持5分钟。然后,在施加4.9mpa压力的状态下,以10℃/分钟的速度缓慢地进行冷却,温度降至室温附近时,从模具中取出成型板。温度23
±
2℃、湿度50
±
5℃的环境下对所得成型板进行48小时以上的状态调节。从状态调节后的压制板冲裁出astm d1822type
‑
s的形状的试验片,作为拉伸冲击强度试验样品。
[0257]
2)拉伸冲击强度试验条件
[0258]
使用上述试验片,参考jis k 7160
‑
1996的b法测定拉伸冲击强度。需要说明的是,与jis k 7160
‑
1996的不同之处仅在于试验片的形状。对于其它测定条件等,采用基于jis k 7160
‑
1996的方法实施试验。
[0259]
(3)复数模量的绝对值g*=0.1mpa下的相位角δ(g*=0.1mpa的测定)
[0260]
1)试样的准备、测定
[0261]
将试样置于厚度1.0mm的热压用模具中,在表面温度180℃的热压机中预热5分钟后,重复进行加压和减压,使熔融树脂中的残留气体脱气,进一步地以4.9mpa进行加压,保持5分钟。然后,移至表面温度25℃的压制机,在4.9mpa的压力下保持3分钟来使其冷却,制作由厚度为约1.0mm的试样形成的压制板。由试样形成的压制板加工为直径25mm的圆形,将其作为样品,作为动态粘弹性特性的测定装置,使用rheometrics公司制ares型旋转式流变仪,在氮气气氛下采用以下条件测定动态粘弹性。
[0262]
·
板:φ25mm(直径)平行板
[0263]
·
温度:160℃
[0264]
·
应变量:10%
[0265]
·
测定角频率范围:1.0
×
10
‑2~1.0
×
102rad/s
[0266]
·
测定间隔:5点/decade
[0267]
相对于复数模量的绝对值g*(pa)的常用对数logg*绘制相位角δ,将相当于logg*=5.0的点的δ(度)的值设为δ(g*=0.1mpa)。当测定点中没有相当于logg*=5.0的点时,使用logg*=5.0前后的2个点,利用线性插值求出logg*=5.0的δ值。另外,测定点均为logg*<5时,从logg*值大的一侧取3个点的值,利用2次曲线外推求出logg*=5.0的δ值。
[0268]
(4)熔点及结晶度
[0269]
熔点由通过差示扫描量热计(dsc)测定的吸热曲线的峰温度来表示。使用sii nanotechnology inc.制的dsc(dsc7020),在以下测定条件下进行测定。
[0270]
将约5.0mg试样装入铝盘中,以10℃/分钟升温至200℃,在200℃下保持5分钟后,以10℃/分钟降温至30℃。在30℃下保持5分钟后,再次以10℃/分钟进行升温时的吸收曲线中,将最大峰温度设为熔点tm,由熔解吸热峰面积求出熔解热(δh),将该熔解热除以高密度聚乙烯(hdpe)的完全结晶的熔解热293j/g,由此求出结晶度(%)。
[0271]
(5)源自羧基和/或二羧酸酐基团单体的结构单元和每1000个碳的分支数的测定方法
[0272]
共聚物中的源自羧基和/或二羧酸酐基团单体的结构单元及每1000个碳的分支数可使用
13
c
‑
nmr谱求出。
13
c
‑
nmr通过以下方法进行测定。
[0273]
将试样200~300mg与邻二氯苯(c6h4cl2)和氘代溴苯(c6d5br)的混合溶剂(c6h4cl2/c6d5br=2/1(体积比))2.4ml以及作为化学位移的标准物质的六甲基二硅氧烷一起放入内径10mmφ的nmr试样管中,进行氮气置换后封管,进行加热溶解制成均匀的溶液作为nmr测定试样。
[0274]
nmr测定使用装有10mmφ冷冻探针的bruker japan株式会社的av400m型nmr装置在120℃下进行。
[0275]
13
c
‑
nmr在试样温度120℃、脉冲角90
°
、脉冲间隔51.5秒、累计次数512次以上的条件下使用反转门控去耦法进行测定。
[0276]
化学位移是将六甲基二硅氧烷的
13
c信号设定为1.98ppm,其它基于
13
c的信号的化学位移以其为基准。
[0277]
1)试样的预处理
[0278]
试样中含有羧酸盐基的情况下,通过进行酸处理将羧酸盐基改性为羧基后,用于测定。试样中含有羧基的情况下,可适当进行例如使用重氮甲烷或三甲基甲硅烷基(tms)重氮甲烷等的甲酯化等酯化处理。
[0279]
2)源自具有羧基和/或二羧酸酐基团的单体的结构单元的计算
[0280]
<e/tba>
[0281]
在
13
c
‑
nmr谱的79.6~78.8处检测到tba的丙烯酸叔丁酯基团的季碳信号。使用该信号强度,由下式算出共聚单体量。
[0282]
tba总量(mol%)=i(tba)
×
100/〔i(tba) i(e)〕
[0283]
其中,i(tba)、i(e)分别为下式所示的量。
[0284]
i(tba)=i
79.6~78.8
[0285]
i(e)=(i
180.0~135.0
i
120.0~5.0
‑
i(tba)
×
7)/2
[0286]
<e/tba/iba>
[0287]
在
13
c
‑
nmr谱的79.6~78.8ppm处检测到tba的丙烯酸叔丁酯基团的季碳信号,在70.5~69.8ppm处检测到iba的异丁氧基的亚甲基信号,在19.5~18.9ppm处检测到异丁氧基的甲基信号。使用该信号强度,由下式算出共聚单体量。
[0288]
tba总量(mol%)=i(tba)
×
100/〔i(tba) i(iba) i(e)〕
[0289]
iba总量(mol%)=i(iba)
×
100/〔i(tba) i(iba) i(e)〕
[0290]
其中,i(tba)、i(iba)、i(e)分别为下式所示的量。
[0291]
i(tba)=i
79.6~78.8
[0292]
i(iba)=(i
70.5~69.8
i
19.5~18.9
)/3
[0293]
i(e)=(i
180.0~135.0
i
120.0~5.0
‑
i(iba)
×7‑
i(tba)
×
7)/2
[0294]
<e/tba/nb>
[0295]
在
13
c
‑
nmr谱的79.6~78.8ppm处检测到tba的丙烯酸叔丁酯基团的季碳信号,在41.9~41.1ppm处检测到nb的次甲基碳信号。使用该信号强度,由下式算出共聚单体量。
[0296]
tba总量(mol%)=i(tba)
×
100/〔i(tba) i(nb) i(e)〕
[0297]
nb总量(mol%)=i(nb)
×
100/〔i(tba) i(nb) i(e)〕
[0298]
其中,i(tba)、i(nb)、i(e)分别为下式所示的量。
[0299]
i(tba)=i
79.6~78.8
[0300]
i(nb)=(i
41.9~41.1
)/2
[0301]
i(e)=(i
180.0~135.0
i
120.0~5.0
‑
i(nb)
×7‑
i(tba)
×
7)/2
[0302]
需要说明的是,各单体的结构单元量由包括不等号的“<0.1”表示时,是指虽然作为多元共聚物中的构成单元存在但考虑到有效数字为小于0.1mol%的量。
[0303]
3)每1000个碳的分支数的计算
[0304]
多元共聚物中存在分支时,有主链中分支独立存在的孤立型、以及复合型(分支与分支隔着主链相对的相对型、支链中有分支的支链分支(branched
‑
branch)型、以及连锁型)。
[0305]
以下为乙基分支的结构的例子。需要说明的是,相对型的例子中,r表示烷基。
[0306][0307]
每1000个碳的分支数通过在下式的i(分支)中代入下述的i(b1)、i(b2)、i(b4)中的任一者来求出。b1表示甲基分支,b2表示乙基分支,b4表示丁基分支。甲基分支数使用i(b1),乙基分支数使用i(b2),丁基分支数使用i(b4)来求出。
[0308]
分支数(个/每1000个碳)=i(分支)
×
1000/i(总数)
[0309]
其中,i(总数)、i(b1)、i(b2)、i(b4)为下式所示的量。
[0310]
i(总数)=i
180.0~135.0
i
120.0~5.0
[0311]
i(b1)=(i
20.0~19.8
i
33.2~33.1
i
37.5~37.3
)/4
[0312]
i(b2)=i
8.6~7.6
i
11.8~10.5
[0313]
i(b4)=i
14.3~13.7
‑
i
32.2~32.0
[0314]
其中,i表示积分强度,i的下角标的数值表示化学位移的范围。例如i
180.0~135.0
表示在180.0ppm与135.0ppm之间检测出的
13
c信号的积分强度。
[0315]
对于归属,参考非专利文献macromolecules 1984,17,1756
‑
1761、macromolecules 1979,12,41。
[0316]
需要说明的是,各分支数由包括不等号的“<0.1”表示时,是指虽然作为多元共聚物中的构成单元存在但考虑到有效数字为小于0.1mol%的量。另外,not detected意为低于检测极限。
[0317]
<(制造例1):b
‑
27dm/ni络合物的合成>
[0318]
b
‑
27dm/ni络合物按照国际公开第2010/050256号记载的合成例4,使用下述的2
‑
双(2,6
‑
二甲氧基苯基)膦基
‑6‑
五氟苯基苯酚配体(b
‑
27dm)。依据国际公开第2010/050256号的实施例1,使用双(1,5
‑
环辛二烯)镍(0)(称为ni(cod)2),合成b
‑
27dm和ni(cod)2以1:1反应的镍络合物(b
‑
27dm/ni)。
[0319][0320]
<(制造例2、制造例3、制造例10、制造例11):离聚物前体树脂用共聚物的制造>
[0321]
使用制造例1中制造的过渡金属络合物(b
‑
27dm/ni络合物),制造乙烯/丙烯酸t
‑
bu/2
‑
降冰片烯(制造例2)、乙烯/丙烯酸t
‑
bu/丙烯酸i
‑
bu(制造例3)、乙烯/丙烯酸t
‑
bu共聚物(制造例10、制造例11)。参考日本特开2016
‑
79408号公报记载的制造例1或制造例3,进行共聚物的制造。金属催化剂种类、金属催化剂量、三辛基铝(tnoa)量、甲苯量、共聚单体种类、共聚单体量、乙烯分压、聚合温度、聚合时间等适当变更的制造条件、制造结果示于表1,所得共聚物的物性示于表2。
[0322]
另外,将制造例2及3的树脂组成代入共聚物的无规性的相关公式中,分别得到100.08、101.20的值。另外,将制造例10及制造例11的树脂组成代入共聚物的无规性的相关公式中,分别得到116.9、116.4的值。这些值为大于所得树脂的熔点的值,满足上述关系式50<tm<
‑
3.74
×
[z] 130,因此可以判断制造例2及3的树脂均为无规性高的树脂。
[0323]
[表1]
[0324][0325]
[表2]
[0326][0327]
<(制造例4、制造例5、制造例12、制造例13):离聚物前体树脂的制造>
[0328]
在内容积1.6m3的带有搅拌叶片的sus316l制高压釜中,加入得到的制造例2、制造例3、制造例10、制造例11的树脂中的任1种100kg、对甲苯磺酸一水合物2.0kg、甲苯173l,在
105℃下搅拌4小时。加入离子交换水173l并进行搅拌,静置后,取出水层。之后,重复进行离子交换水的加入和取出直至取出的水层的ph变为5以上。将剩余的溶液加入42mmφ带排气装置的双螺杆挤出机(l/d=42)中,通过排气口抽真空来蒸馏去除溶剂。进而,将由挤出机前端的模头连续地以线料形式挤出的树脂在水中进行冷却,用切割机切断,由此得到树脂的粒料。
[0329]
所得树脂的ir谱中,观测到源自t
‑
bu基的850cm
‑1附近的峰的消失以及源自酯的羰基的1730cm
‑1附近的峰的减少、源自羧酸(二聚体)的羰基的1700cm
‑1附近的峰的增加。由此,确认t
‑
bu酯的分解及羧酸的生成,得到制造例4、制造例5、制造例12、制造例13。所得前体树脂的物性示于表3。
[0330]
[表3]
[0331][0332]
<(制造例6~制造例9、制造例14~制造例16)离聚物的制造>
[0333]
1)na离子供给源的制造
[0334]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:roller mixerr60型中,加入乙烯/甲基丙烯酸(maa)共聚物(dow
‑
mitsui polychemicals co.,ltd.制商品名:nucrel n1050h)22g和碳酸钠18g,在180℃下以40rpm混炼3分钟,由此制造na离子供给源。
[0335]
2)zn离子供给源的制造
[0336]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:roller mixerr60型中,加入乙烯/甲基丙烯酸(maa)共聚物(dow
‑
mitsui polychemicals co.,ltd.制商品名:nucrel n1050h)21.8g和氧化锌18g、硬脂酸锌0.2g,在180℃下以40rpm混炼3分钟,由此制造zn离子供给源。
[0337]
3):离聚物的制造
[0338]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:roller mixerr60型中,加入离聚物前体树脂40g,在160℃下以40rpm混炼3分钟,使其溶解。然后,加入离子供给源以形成期望的中和度,在250℃下以40rpm混炼5分钟。制造离聚物时,离聚物前体树脂的种类选自制造例4及制造例5、制造例12及制造例13,并且作为离子供给源分别组合na和zn,制作制造例6~制造例9及制造例14~制造例16的离聚物。确认各制造例中得到的离聚物的ir谱,可知源自羧酸(二聚体)的羰基的1700cm
‑1附近的峰减少、源自羧酸盐基的羰基的1560cm
‑1附近的峰增加。所得离聚物的物性示于表4。
[0339]
〔实施例1/聚酰胺树脂组合物的制备〕
[0340]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:
roller mixerr60型中,加入聚酰胺(东丽株式会社制,商品名:amilan(注册商标),等级:cm6246m)树脂44g及制造例6的离聚物11g,在230℃下以50rpm混炼5分钟。从混炼机中取出树脂组合物,并进行冷却,由此得到实施例1的树脂组合物。
[0341]
〔实施例2~8、比较例1~6、实施例9~15、比较例8、比较例9/聚酰胺树脂组合物的制备〕
[0342]
将聚酰胺树脂的种类和量、离聚物的种类和量、离聚物前体树脂的种类和量分别变更为表5、6中记载的内容,除此以外,利用与实施例1相同的方法得到实施例2~8、实施例9~15及比较例1~6、比较例8、比较例9的树脂组合物。需要说明的是,表5、6中记载的him1605、him1707、him1555、him1707使用各等级的dow
‑
mitsui polychemicals co.,ltd.制商品名:himilan。另外,实施例12~实施例15中使用聚酰胺12(ems
‑
chemie holding ag制商品名:grilamid l25ah)。表中的no data表示无资料、不详。
[0343]
〔比较例7〕
[0344]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:roller mixerr60型中,仅加入聚酰胺(东丽株式会社制,商品名:amilan(注册商标),等级:cm6246m)55g,在230℃下以50rpm混炼5分钟。从混炼机中取出树脂组合物,并进行冷却,由此得到比较例7的树脂组合物。
[0345]
〔比较例10〕
[0346]
在容量60ml的安装有小型混合器的东洋精机株式会社制labo plastomill:roller mixerr60型中,仅加入聚酰胺12(ems
‑
chemie holding ag制商品名:grilamid l25ah)55g,在230℃下以50rpm混炼5分钟。从混炼机中取出树脂组合物,并进行冷却,由此得到比较例7的树脂组合物。
[0347]
[表4]
[0348][0349]
[表5]
[0350][0351]
[表6]
[0352][0353]
4.实施例/比较例的研究
[0354]
实施例1~实施例11为满足本发明的技术特征的聚酰胺树脂组合物,表现出高耐冲击性。实施例3~实施例8的聚酰胺树脂组合物在各种树脂组成比率下分别表现出优异的耐冲击性,表明无论离聚物的添加量如何,均具有耐冲击性的改善效果。另一方面,比较例7为聚酰胺树脂单体,因此耐冲击性差。比较例1~比较例4中将共混的离聚物的相位角小于50
°
的不满足本发明的技术特征的、具有包含大量长支链的结构的离聚物与聚酰胺树脂共混,因此耐冲击性的改善不充分。特别是,比较例3表明有时以往的离聚物还会降低耐冲击性。比较例5及比较例6中,将形成离聚物前的前体树脂与聚酰胺树脂共混,因此耐冲击性的改善不充分。
[0355]
实施例12~实施例15为满足本发明的技术特征的聚酰胺树脂组合物,表现出了高耐冲击性。该事实表明,不论聚酰胺树脂的种类,只要是满足本技术的技术特征的聚酰胺树脂组合物就具有耐冲击性的改善效果。另一方面,比较例10为聚酰胺树脂单体,因此耐冲击性差。比较例8、比较例9中,将共混的离聚物的相位角小于50
°
的不满足本发明的技术特征的、具有包含大量长支链的结构的离聚物与聚酰胺树脂共混,因此耐冲击性的改善不充分。
[0356]
由以上各实施例的良好结果及与各比较例的对照可以看出本发明的构成(发明限定内容)的意义和合理性、以及相对于现有技术的卓越性。
[0357]
产业上的可利用性
[0358]
使用本发明离聚物的聚酰胺树脂组合物与以往的树脂相比耐冲击性优异。因此,
本发明特别有效地用于汽车部件、电动工具外壳、机械部件、电气/电子部件、家庭/办公用品、建材相关部件、家具用部件、包装材料、高尔夫球等各种成型品。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。