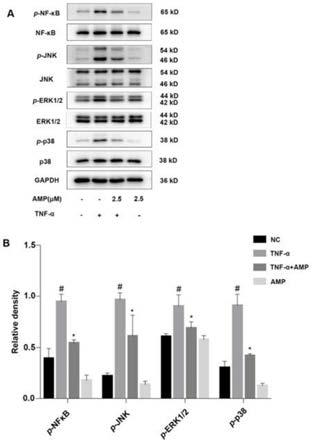
1.本发明涉及基于多个基因组比对的参考基因组及其注释更新、基于二代测序数据的序列结构变异的挖掘、基于多样本基因组结构变异的群体基因型构建以及基于结构变异基因型的全基因组关联分析方法,特别涉及一种基于多个基因组比较和二代测序数据的全基因组关联分析方法,属于生物信息技术领域。
背景技术:
2.全基因组关联分析是对多个个体在全基因组范围的遗传变异多态性进行检测,获得基因型,进而将基因型与可观测的表型进行群体水平的统计学分析,挖掘与性状变异相关的基因。全基因组范围的遗传变异形式多样,有碱基置换变异、插入、缺失、倒位易位等。目前随着二代测序成本降低,基于二代测序技术挖掘基因组遗传变异、构建群体基因型进行全基因组关联分析的研究越来越多。基于二代测序数据挖掘基因组遗传变异需要参考基因组和测序数据,为了获得更丰富更准确的遗传变异,可从参考基因组、样品测序数据和遗传变异检测方法三方面改进:1.为了获得更有利的参考基因组,有研究者提出使用泛基因组作为参考基因组,泛基因组是指群体中全部的基因组序列的总和。通过捕获、呈现群体中全部的基因组变异,为功能基因组学研究提供完整的参考基因组。但是目前构建泛基因组策略有很多不足之处,泛基因组的组织形式也无法统一,由于基因组变异种类多样,表明泛基因组或许不应是线性的,有研究者提出基于图论的基因组,但是多维图形式的泛基因组不容易理解,并且现有的二代测序数据比对到参考基因组上的经典分析软件如bwa也只适用于线性的参考基因组;2.全基因组测序数据方面,相对于三代测序技术、hi
‑
c技术等,二代测序技术在价格和准确性存在优势,可提供挖掘群体全基因组结构变异的有效测序数据;3.目前利用二代测序数据和线性参考基因组检测结构变异的方法多样,包括依靠测序覆盖深度方法、配对双末端测序方法、序列读长分割的方法和组装的方法,各类方法各有特色,优势劣势并存。本发明创新性利用一种新的策略,把基因组比较得到的结构变异重新整合到参考基因组上,多次迭代获得更新后的参考基因组及其注释,使用比对软件将二代测序数据挂载到参考基因组上,根据覆盖深度重新挖掘序列结构变异并构建群体基因组结构变异基因型用于全基因组关联分析。
技术实现要素:
3.本发明的目的在于提供一种基于多个基因组比较和二代测序数据的全基因组关联分析方法,以解决上述背景技术中提出的问题。
4.为实现上述目的,本发明提供如下技术方案:一种基于多个基因组比较和二代测序数据的全基因组关联分析方法,所述分析方法步骤如下:
5.步骤一:使用比对软件将参考基因组和从头组装基因组文件进行比对,获取基因组间的共线性特征,根据共线性特征挖掘结构变异位点,将从头组装基因组中的插入片段
插入至参考基因组相应位置中,生成更新后的参考基因组文件;
6.步骤二:根据插入片段大小及插入到参考基因组的位置,更新参考基因组中的注释基因位置或结构,根据插入片段位置与从头组装基因组中基因注释信息,获得基因结构完全位于插入片段的注释基因并更新到参考基因组中;
7.步骤三:如果有多个从头组装基因组,依次迭代更新参考基因组;
8.步骤四:利用比对软件将样品的二代测序数据比对到更新后的参考基因组上,利用序列提取软件提取比对文件的短序列覆盖度,生成覆盖度文件,通过设定覆盖度阈值的方式,判断样品的结构变异情况;
9.步骤五,收集所有样品全部结构变异的分界点位置信息至一个集合中,利用分界点位置将更新后的参考基因组分割成一个个片段(命名为bin),重新捕获所有bin在样品中的存在与缺失情况,构建出每个样品基于bin的基因型,将所有样品的bin基因型进行合并,经过最小等位基因频率(maf)筛选,形成群体基因组结构变异基因型;
10.步骤六:基于群体基因组结构变异基因型和表型进行全基因组关联分析,根据关联位点和更新后的参考基因组注释文件进行功能基因的候选。
11.作为本发明的一种优选技术方案,所述步骤一将检测到的来自应比对基因组的片段插入到参考基因组相应位置中,指大于50bp的片段。
12.作为本发明的一种优选技术方案,所述结构变异指序列片段存在和缺失。
13.作为本发明的一种优选技术方案,所述步骤五将所有样品的结构变异的分界点位置信息收集到集合中,集合的中的元素将参考基因组分割成bin,bin的划分有利于所有样品标记数量和位置的统一,从而实现群体基因型构建。
14.与现有技术相比,本发明的有益效果是:
15.1.本发明一种基于多个基因组比较和二代测序数据的全基因组关联分析方法,本发明实现了利用结构变异基因型数据进行全基因组关联分析。
16.2.本发明创新性将多个基因组整合,更新参考基因组,更新后的参考基因组包含更多初始参考基因组不存在的片段,为后续检测结构变异和挖掘新的功能基因提供了可能。并且更新后的参考基因组依然为线性形式,方便理解和后续应用。
17.3.本发明利用二代测序比对到更新后的参考基因组上,通过覆盖度获取结样品的结构变异信息,实现了高效、精准的基因组结构变异分析和鉴定。
18.4.本发明创新性构建了bin图谱,针对每个样品结构变异起止位置和大小不一样的情况,统一了所有样品结构变异数量和位置,形成基因型文件用于全基因组关联分析。由于结构变异普遍存在并可能直接导致基因功能的改变,因此利用结构变异基因型进行全基因组关联分析,相对于snp基因型进行全基因组关联分析可找到新的关联位点。
19.5.本发明创新性更新了参考基因组的基因注释,通过关联位点位置和更新后的基因注释可以进行基因候选,并且由于更新后的基因注释还包含部分来自从头组装基因组的基因,因此可能找到初始参考基因组上不存在的功能基因,这是利用初始参考基因组进行基因候选无法实现的。
附图说明
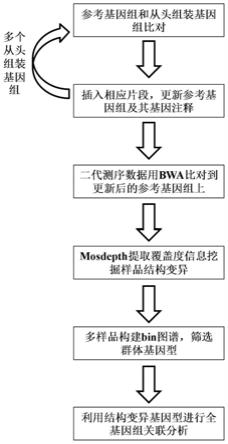
20.图1为本发明的主要流程示意图;
21.图2为本发明的参考基因组的更新示意图;
22.图3为本发明的参考基因组基因注释的更新示意图;
23.图4为本发明的单个样品结构变异检测新示意图;
24.图5为本发明的群体基因型构建示意图;
25.图6为本发明的水稻抗病全基因组关联分析。
具体实施方式
26.对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
27.请参阅图1
‑
6,本发明提供了一种基于多个基因组比较和二代测序数据的全基因组关联分析方法:
28.a.参考基因组及其注释的更新:
29.a1.将参考基因组和多个从头组装基因组名称保存到一个文本文件中并排序,其中第一行为参考基因组,第二行及后续行设为从头组装基因组。
30.a2.使用比对软件将参考基因组和从头组装基因组文件进行比对,获取基因组间的共线性特征。
31.a3.使用提取软件根据步骤a2获取的共线性特征进行结构变异位点挖掘,以步骤a2的参考基因组为位置基准,生成结构变异位点位置信息文件。
32.a4.根据步骤a3产生的结构变异位置信息文件,将只存在于从头组装基因组中的片段插入至参考基因组相应位置中,生成新的参考基因组文件。
33.a5.根据插入片段大小及插入到参考基因组的位置,更新参考基因组中的注释基因位置或结构。
34.a6.根据插入片段位置与从头组装基因组中基因注释信息,获得基因结构完全位于插入片段的注释基因。将此类注释基因加入a5生成的基因注释文件中,更新参考基因组的基因注释。
35.a7.如果存在多个从头组装基因组,则步骤a4中更新后的参考基因组文件作为参考基因组,步骤a6中生成的基因注释文件作为参考基因组的基因注释。结合a1文本文件的第三行及后续行的从头组装基因组依次重复步骤a2
‑
a6,最终生成多次迭代更新后的参考基因组文件及其基因注释文件。
36.b.基于更新后参考基因组与二代测序数据的全基因组结构变异挖掘:
37.b1.通过比对软件将样品的二代测序数据比对到步骤a中更新后的参考基因组上,生成比对文件。
38.b2.利用序列提取软件对比对文件的短序列覆盖度进行提取,生成覆盖度文件。通过设定覆盖度阈值的方式,判断样品的结构变异情况。
39.c.群体基因组结构变异基因型的构建:
40.c1.收集所有样品全部结构变异的起始终止位点信息去除冗余,放入一个集合中,集合中的位点可将参考基因组切割成不同大小的片段(简称bin,bin用bin起始位置命名)。
41.c2.根据样品的结构变异情况,重新捕获bin在样品中的存在与缺失情况,构建出
每个样品基于bin的基因型。
42.c3.将所有样品的bin基因型进行合并,经过最小等位基因频率(maf)筛选,形成群体基因组结构变异基因型。
43.d.使用群体基因组结构变异基因型和表型进行全基因组关联分析,根据关联位点位置在更新后的参考基因组基因注释文件中寻找候选基因。
44.注:
45.1.本发明实现步骤a2中使用比对软件为mummer,步骤a3使用提取软件为assemblytics,步骤b1中使用比对软件为bwa,步骤b2中使用序列提取软件为mosdepth。
46.2.mummer,assemblytics,bwa,mosdepth为软件且均无中文名称,在行业内直接用英文表达。
47.3.本发明b2步骤中覆盖度阈值为用户设定,若变异位点二代测序数据比对覆盖度大于阈值,则说明序列比对覆盖此变异片段,该样品存在此处变异位点,反之,则不存在。
48.4.本发明b2步骤中可设定最小缺失(gap)阈值,若检测到的变异大于该阈值,该样品存在此处变异位点,反之,则不存在。
49.5.本发明的结构变异指dna序列的存在与缺失。
50.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
51.下面结合附图对本发明的应用原理作详细的描述。本发明共包含三个部分。
52.实施例一:水稻参考基因组及其基因注释更新
53.使用已经组装和注释的33个水稻基因组,以水稻日本晴msu作为初始参考基因组,依次比对dhx2,02428,kosh,zh11,ky131,lemont,namroo,lj,g46,cn1,fs32,dg,d62,ii32,r527,s548,9311,y58s,j4155,g8,y3551,ir64,r498,tm,tumba,g630,yx1,wssm,fh838,n22,basmati1,cg14。
54.如图2所示,第一轮比对中,利用mummer软件将参考基因组(msu)和第一个从头组装基因组(dhx2)进行比对,获得基因组间的共线性特征,生成delta文件;
55.利用assemblytics软件提取共线性特征,利用python软件整理共线性特征文件筛选插入片段大小(>50bp),获得以参考基因组为位置基准的结构变异位置信息数据文件(assemblytics_structural_variants.bed文件),该bed包含比对的位置信息;
56.运行awk对bed文件进行筛选,得到coords.bed,只包含插入和缺失片段;
57.运行3replace_regions_with_nucleotides1.2.py程序根据bed位置和dhx2基因组序列调取插入片段序列;
58.运行grep对bed大小进行筛选,去除插入片段小于50bp的片段;
59.运行自编程序5ins.bed_to_bed2.py,对bed进行格式整理,形成bed2格式文件,该文件格式为:第一列为插入片段对应在msu基因组上的染色体,第二列为插入片段对应在msu基因组上的起始位置,第三列为第二列数据加上插入片段大小之和,第四列为“insertion”标签,第五列为插入碱基序列,第六列为dhx2基因组名称,第七列为插入片段在dhx2基因组上的染色体,第八列为插入片段在dhx2的起始位置。
60.运行自编程序6update_ref_by_nucmer.py,根据msu基因组序列、dhx2基因组序
列,和比对结果bed2格式文件,将来自dhx2基因组的序列插入到msu基因组的相应位置中,获得更新后的参考基因组ref0dhx2.fa。
61.如图3所示,所有从头组装基因组和参考基因组包含基因组序列(fa)文件和基因注释(gff)文件。当参考基因组插入来自从头组装基因组的序列片段(presence variation,pv)时,通过pv在从头组装基因组的位置与大小和从头组装基因组基因注释信息获取注释基因与pv的重叠情况,筛选出完全位于pv上的基因;根据pv插入到参考基因组的位置和参考基因组基因注释获取插入位点与基因的重叠情况,筛选位于基因内部的插入位点。利用r语言软件根据位置信息更新基因注释,包含以下情况:
62.1.若pv位于基因位置前,基因的位置应在原有位置加上pv大小,得到新的位置信息,以保证新的基因注释gff文件与新的参考基因组相对应;
63.2.若pv位于基因内部,pv前段基因结构注释情况不变,pv后段基因结构注释应加上pv大小,得到新位置信息;
64.3.若pv内包含完整基因结构,应将新带入的基因纳入参考基因组基因注释中。更新之后得到一个与更新后的参考基因组相对应的基因注释(gff)文件。
65.运行自编程序7bed2_update_bed3.r,根据bed2文件获得bed3文件,bed3与bed2格式一致,但是bed3里的前三列为插入片段对应在ref0dhx2.fa的染色体和起始终止位置。
66.运行自编程序8gff_update_by_bed2info_parlapply.r,根据msu.gff注释文件和bed2文件更新注释基因位置,其中部分基因可能包含插入片段,此步骤只更新原有位置信息,获得ref0.update1.gff。
67.运行自编程序9gff_split_by_bed3_6.r,将被插入片段打断的基因注释更新,记录被打断状态,获得ref0.update2.gff。
68.运行自编程序10gene_in_pv_from_gff_parlapply2.r,根据bed3和dhx2基因注释获得完全位于插入片段上的gene、mrna、exon和cds等注释信息,得到dhx2.gff.in_pv文件。
69.运行自编程序11gene_in_pv_screen.py筛选dhx2.gff.in_pv,去除部分exon或cds在插入片段上而基因不完全在插入片段的注释信息。得到基因完全位于插入片段的注释信息,生成dhx2.gff_gene_absolutly.in_pv文件。
70.运行自编程序12bed3_update_gff.r,根据bed3文件和dhx2.gff_gene_absolutly.in_pv文件,得到插入片段基因位于更新后参考基因组ref0dhx2.fa上的位置,生成hx2.gene_absolutly_in_pv.gff注释文件。
71.将ref0.update2.gff和hx2.gene_absolutly_in_pv.gff合并,获得与ref0dhx2.fa相对应的基因注释文件ref0dhx2.gff。
72.初始参考基因组msu大小为356mb,经过与dhx2进行比对、提取共线特征、插入序列等操作后,更新后的参考基因组大小为359mb,增加约了3mb的碱基序列。基因注释gff中增加了87个基因。
73.以ref0dhx2.fa创建软链接为ref1.fa,ref0dhx2.gff创建软链接为ref1.gff,以ref1为参考基因组与下一个从头组装基因组02428进行比对、提取共线特征、插入序列等操作得到第二次更新后的参考基因组及其基因注释,依次迭代更新,获得多轮迭代更新后的参考基因组及其基因注释。本实施例共33个基因组,经过32轮更新后得到基因组ref32.fa和ref32.gff。ref32.fa大小为428mb,比msu大72mb,ref32.gff包含58235个基因,比msu多
出2249个基因。使用gffread软件,根据ref32.fa和ref32.gff提取基因组对应的所有蛋白序列,得到ref32的蛋白序列文件,同样方法获得msu的蛋白序列文件。msu的12条染色体上存在66153个蛋白序列,根据msu的蛋白序列id在ref32的蛋白序列文件中进行搜索,结果表明:相同id在两个蛋白文件中对应的蛋白序列完全相同。这表明基因注释更新的准确性。
74.实施例三:基于二代测序数据与更新后的参考基因组的序列结构变异挖掘
75.如图4所示,利用bwa文件将二代测序数据(fq)挂载到更新后的参考基因组上,输出数据利用管道和samtools软件转换成bam文件,使用picad软件对bam文件进行排序,去除重复操作得到sorted_add_dedup.bam文件,利用samtools是软件将比对质量小于20、挂载不上参考基因组、匹配到多处的测序片段去除,得到筛选后的mapq20.bam文件。
76.利用自编程序2map_fq_to_pan.py将fqd_dir目录下的所有测序数据挂载带参考基因组上,生产bam_dir文件夹,包含所有样品的mapq20.bam文件。测序数据fq.gz文件放在同一个目录fqd_dir下,双端测序数据格式要统一,本实例中为_1.fq.gz和_2.fq.gz为后缀。命令为python32map_fq_to_pan.py
‑
fqd fq_dir
‑
r ref32.fa
‑
br bam_dir
77.利用mosdepth软件提取mapq20.bam文件的覆盖度信息得到per
‑
base.bed.gz文件,为了减少后续计算量,我们使用20为窗口,计算每20个碱基区域内的覆盖度,结果文件保存在regions.bed.gz内。本实施例共414个二代测序数据,其中所有样品per
‑
base.bed.gz文件的平均行数为2,104,638行,所有样品regions.bed.gz文件的平均行数为601,375行。
78.运行自编程序1depth_call_pav.py,从regions.bed.gz提取覆盖度信息,设置覆盖度阈值为5,将覆盖度小于阈值的区域判定为缺失,否则判定为存在。将小于50bp的插入缺失忽略,记录存在和缺失的交界点,所有序列存在和缺失信息记录在5depth_50bp.txt文本文件中。本实施例414个样品,平均每个样品包含85,823个插入片段。
79.实施例三:群体基因组结构变异基因型构建
80.如图5所示,利用r语言软件提取群体中所有样品的存在和缺失交界点位置信息,去除冗余放入到集合中。所有交界点将参考基因组分割成若干大小的片段区间(称为bin),每个bin使用染色体加片段左侧位置进行命名,从记录每个样品的序列存在和缺失信息的per
‑
base.bed.gz文件中,逐个分析出每个bin在每个样品的存在和缺失状态。利用r语言软件将群体的存在缺失状态记录成矩阵形式,一个bin为一个遗传标记,该bin图谱中,每行代表一个样品,每列代表一个bin标记。
81.运行自编程序2merge_samples_pav_to_population.r,可输出所有标记位置文件all_chr_pos_frame.txt和bin图谱文件。本实施例all_chr_pos_frame.txt包含6,270,114个标记。
82.运行自编程序3pav_genotype_to_hmp.py,对bin图谱的每一列进行筛选,去除bin标记基因型全为缺失状态或全为存在状态列,再筛选最小等位基因频率(maf),转换成hapmap格式的群体基因组结构变异基因型。本实例中包含414个国际稻种资源,筛选后的基因型包含4,756,248个bin标记。
83.实施例四:全基因组关联分析
84.使用gapit软件进行全基因组关联分析。
85.利用初始参考基因组和测序数据,利用bwa和gatk软件获取群体snp基因型,使用
gapit软件进行全基因组关联分析。
86.如图6所示比较结构变异基因型和snp基因型全基因组关联分析结果,可以发现,使用snp基因型能找到的关联位点,用结构变异基因型几乎都能找到,并且结构变异基因型还能找到新的关联位点。由于更新后的基因注释还带入了新的基因,弥补了初始参考基因组注释基因数量有限的缺点。
87.尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。