1.本发明涉及利用合成领域,具体涉及一种人工烟酰胺辅因子介导的酶法降解二苯并噻吩的方法。
背景技术:
2.硫在原油中的平均含量在0.05%
‑
5%之间,在重油中的含量甚至可以高达14%,是含量仅次于c和h的第三大元素。原油中70%以上的硫以二苯并噻吩及其衍生物的形式存在,这些化合物在环境中可以稳定存在三年以上。随着燃油泄漏,具有致突变性的二苯并噻吩释放到环境中,生态系统中生物造成很大影响。生物脱硫因其费用低、条件温和、利于环保,而且对传统的加氢脱硫难以去除的二苯并噻吩等化合物具有很好的脱除效果而受到人们的重视。由于二苯并噻吩及其衍生物是原油中硫的主要存在形式,因而生物脱硫通常以dbt作为模式化合物。目前报道的微生物代谢有机硫的途径有三条,碳碳键裂解途径、硫氧化途径和硫专一性代谢途径。
3.碳碳键裂解途径和硫氧化途径是细菌的代谢途径,而不是脱硫途径。这两条途径破坏碳骨架而损失了燃烧值,因而其对于石油及其产品的生物脱硫并不具有实际应用价值。硫专一性代谢途径能够专一性脱除有机硫并且不会因破坏碳骨架而损失燃烧值。在硫专一性代谢途径中,涉及到一种黄素依赖性双组份单加氧酶
‑
二苯并噻吩单加氧酶,这种酶由黄素还原酶dszd和加氧酶dszc组成。在催化反应的过程中需要用到烟酰胺辅因子和黄素fmn。
4.烟酰胺辅因子在催化过程中会与底物发生化学反应,并转化为相应的还原或氧化形式,因为不能再生,将其量化并用于实际的大规模合成的成本太高。目前解决这一问题主要是实现辅因子的再生或者合成廉价的人工辅因子来取代天然辅因子。合成人工辅因子的技术已经相对比较成熟,在和酶的结合使用上也有很多前人做过各种各样的尝试,人工烟酰胺辅因子可以代替天然烟酰胺辅因子来充当氢供体。虽然很多人工辅因子转移电子的效率比不上天然的辅因子,但这是有效解决天然辅酶不稳定不经济的手段之一。
5.目前用于辅因子再生的四种方法分别是酶法再生、化学法再生、电化学法再生、光化学法再生。酶法一般用到一个或者多个酶,酶级联反应的效率较低,易发生解耦连。化学法需要有机贵金属配合物来做催化剂,成本高且污染较大。电化学法易导致酶失活,光化学法受限于缺乏高效的光催化剂,效率较低。
技术实现要素:
6.发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种人工烟酰胺辅因子介导的酶法降解二苯并噻吩的方法。
7.发明思路:本发明旨在合成人工烟酰胺辅因子,替代天然的黄素还原酶和昂贵的nadh,为黄素单核苷酸(fmn)供氢生成还原型黄素单核苷酸(fmnh2)来参与后续氧化反应,催化二苯并噻吩单加氧酶氧化二苯并噻吩生成二苯并噻吩砜。
8.为了解决上述技术问题,本发明公开了一种人工烟酰胺辅因子介导的酶法降解二苯并噻吩的方法,二苯并噻吩(dbt)在二苯并噻吩单加氧酶、黄素单核苷酸(fmn)、人工烟酰胺辅因子(mnadhs)和过氧化氢酶催化反应下生成二苯并噻吩砜(dbto2)。
9.其中,所述反应是在氧气氛围下,或空气氛围下反应。
10.其中,如图2所示,所述人工烟酰胺辅因子还原fmn,生成fmnh2,参与下一步氧化酶催化的氧化底物反应(dbt氧化为dbto2)。
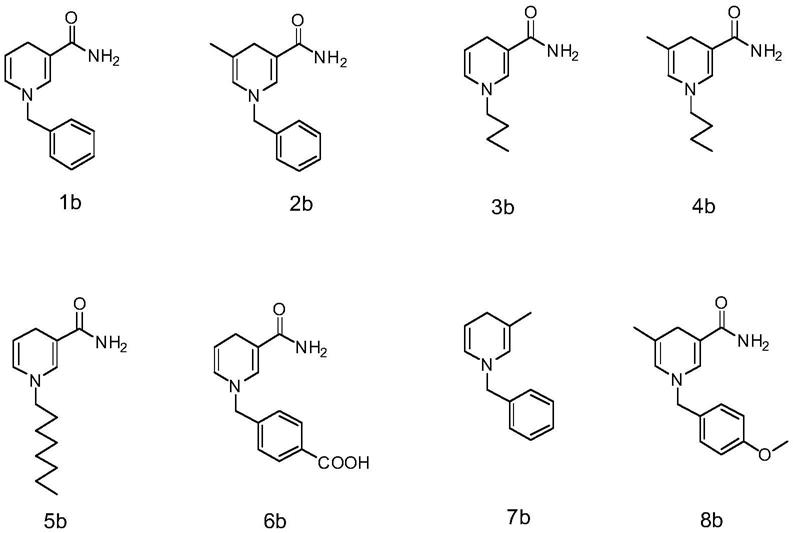
11.其中,所述人工烟酰胺辅因子如式i所示:
[0012][0013]
其中,
[0014]
r1选自氢或甲基;
[0015]
r2选自
‑
conh2、氢或甲基;
[0016]
r3选自苄基、正丁基、正辛基、对甲基苯甲酸酯基或对甲氧基苯甲基。
[0017]
优选地,所述人工烟酰胺辅因子为1
‑
苄基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(1b)、1
‑
苄基
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(2b)、1
‑
丁基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(3b)、1
‑
丁基
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(4b)、1
‑
辛基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(5b)、4
‑
((3
‑
氨基甲酰基吡啶
‑
1(4h)
‑
基)甲基)苯甲酸(6b)、1
‑
苄基
‑3‑
甲基
‑
1,4
‑
二氢吡啶(7b)和1
‑
(4
‑
甲氧基苄基)
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(8b)中的任意一种或几种组合;进一步优选地,所述人工烟酰胺辅因子为1
‑
丁基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(3b)、1
‑
丁基
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(4b)、1
‑
苄基
‑3‑
甲基
‑
1,4
‑
二氢吡啶(7b)和1
‑
(4
‑
甲氧基苄基)
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺(8b)中的任意一种或几种组合;最优选地,所述人工烟酰胺辅因子为1
‑
苄基
‑3‑
甲基
‑
1,4
‑
二氢吡啶(7b)。
[0018]
[0019]
其中,本发明所述人工烟酰胺辅因子均可参考现有公开的文献自行合成。
[0020]
其中,所述反应体系中以缓冲液为溶剂;优选地,以tris
‑
hcl缓冲液为溶剂;进一步优选地,以ph 7
‑
8的tris
‑
hcl缓冲液为溶剂。
[0021]
其中,反应体系中,所述二苯并噻吩的浓度为0.5
‑
2.0mm;优选地,所述二苯并噻吩的浓度为1mm。
[0022]
其中,反应体系中,所述二苯并噻吩单加氧酶的浓度为1
‑
5u/ml反应体系;优选地,所述二苯并噻吩单加氧酶的浓度为3u/ml反应体系。
[0023]
其中,所述二苯并噻吩单加氧酶为dszc酶(eu527978)和/或dszd酶(eu154996);优选地,所述二苯并噻吩单加氧酶为3u/ml反应体系的dszc酶,或dszc酶3u/ml反应体系、dszd酶1u/ml反应体系;优选地,所述二苯并噻吩单加氧酶为3u/ml反应体系的dszc酶。
[0024]
其中,反应体系中,所述黄素单核苷酸的浓度为5
‑
15μm;优选地,所述黄素单核苷酸的浓度为8
‑
12μm。
[0025]
其中,反应体系中,所述人工烟酰胺辅因子的浓度为2
‑
30mm;优选地,所述人工烟酰胺辅因子的浓度为5
‑
30mm;进一步优选地,所述人工烟酰胺辅因子的浓度为8
‑
15mm。
[0026]
其中,所述过氧化氢酶的用量为20
‑
70u/ml反应体系;优选地,所述过氧化氢酶的用量为30
‑
50u/ml反应体系。
[0027]
其中,反应体系的ph为5
‑
9;优选地,反应体系的ph为7
‑
8。
[0028]
其中,反应的温度为25
‑
40℃;优选地,反应的温度为30
‑
35℃。
[0029]
其中,反应过程中的转速为10
‑
1000rpm;优选地,所述转速为100
‑
500rpm;进一步优选地,所述转速为200rpm。
[0030]
其中,所述反应的时间为2
‑
24h。
[0031]
有益效果:与现有技术相比,本发明具有如下优势:
[0032]
本发明避免使用昂贵的天然辅因子来催化反应,使用容易合成的廉价类烟酰胺辅因子帮助酶催化反应,降低了成本;与天然辅因子相比,人工烟酰胺辅因子更加稳定,适用范围广泛;且人工辅因子7b供氢能力优于天然辅因子,催化反应的转化率在4h之后可达90%以上。
附图说明
[0033]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0034]
图1为mnadhs的合成路线
[0035]
图2为人工辅因子替代天然辅因子催化dbt生成dbto2的反应路线图。
具体实施方式
[0036]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0037]
下述实施例中所述dszc酶、dszd酶和过氧化氢酶(阿拉丁c100456)的比活力分别为15u/mg、20u/mg、3000u/mg。
[0038]
一个dszc酶活力单位(u)定义为1分钟内催化1μmol dbt生成1μmoldbto2所需要的
酶量。
[0039]
一个dszd酶活力单位(u)定义为每分钟催化1μmol nadh生成1μmolnad所需要的酶量。nadh在340nm处的光吸收系数为6.01mmol
‑1l cm
‑1。
[0040]
实施例1以图1所示的方式合成还原型辅因子
[0041]
(1)氧化型辅因子的合成
[0042]
在80ml乙腈溶液中加入烟酰胺(10mmol),然后分别加入溴化苄(12mmol)、1
‑
溴丁烷(12mmol)、1
‑
溴辛烷(12mmol)、4
‑
溴甲基苯甲酸乙酯(12mmol)、4
‑
甲氧基溴苄(12mmol),反应温度为80℃,回流加热搅拌12h左右,反应过程中有白色沉淀析出。反应结束后过滤得到粗产物,乙酸乙酯洗涤(15ml
×
3),真空干燥,产率如下:
[0043]
1a(1
‑
苄基
‑3‑
氨基甲酰基吡啶
‑1‑
鎓盐):94%,
[0044]
3a(1
‑
丁基
‑3‑
氨基甲酰基吡啶
‑1‑
鎓盐):91%,
[0045]
5a(1
‑
辛基
‑3‑
氨基甲酰基吡啶
‑1‑
鎓盐):92%,
[0046]
6a(3
‑
氨基甲酰基
‑1‑
(4
‑
乙氧基
‑4‑
氧丁基)吡啶
‑1‑
鎓盐):45%。
[0047]
在80ml乙腈溶液中加入5
‑
甲基烟酰胺(10mmol),然后分别加入溴化苄(12mmol)、1
‑
溴丁烷(12mmol)、4
‑
甲氧基溴苄(12mmol),反应温度为80℃,回流加热搅拌12h左右,反应过程中有白色沉淀析出。反应结束后过滤得到粗产物,乙酸乙酯洗涤(15ml
×
3),真空干燥,产率如下:
[0048]
2a(1
‑
苄基
‑5‑
甲基
‑3‑
氨基甲酰基吡啶
‑1‑
鎓盐):90%,
[0049]
4a(1
‑
丁基
‑5‑
甲基
‑3‑
氨基甲酰基吡啶
‑1‑
鎓盐):89%,
[0050]
8a(3
‑
氨基甲酰基
‑1‑
(4
‑
甲氧基苄基)
‑5‑
甲基吡啶
‑1‑
鎓盐):70%。
[0051]
在80ml乙腈溶液中加入3
‑
甲基吡啶(10mmol),然后加入溴化苄(12mmol)、,反应温度为80℃,回流加热搅拌12h左右,反应过程中有白色沉淀析出。反应结束后过滤得到粗产物,乙酸乙酯洗涤(15ml
×
3),真空干燥,产率如下:
[0052]
7a(1
‑
苄基
‑3‑
甲基吡啶
‑1‑
鎓盐):93%。
[0053]
(2)还原型辅因子的合成
[0054]
称取2mmol氧化型人工辅因子和碳酸氢钠(8mmol)于茄形烧瓶中,加入25ml超纯水搅拌至溶解,将低亚硫酸钠(8mmol)溶于5ml超纯水,缓慢滴加至反应液中,30min内滴加完。室温条件下氩气保护反应4
‑
6h,有沉淀析出,过滤,超纯水洗涤(15ml
×
3),真空冷冻干燥后得到固体产物,产率如下:
[0055]
1b(1
‑
苄基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):33%,
[0056]
2b(1
‑
苄基
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):30%,
[0057]
3b(1
‑
丁基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):28%,
[0058]
4b(1
‑
丁基
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):31%,
[0059]
5b(1
‑
辛基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):29%,
[0060]
6b(4
‑
((3
‑
氨基甲酰基吡啶
‑
11(4h)
‑
基)甲基)苯甲酸):24%,
[0061]
7b(1
‑
苄基
‑3‑
甲基
‑
1,4
‑
二氢吡啶):31%,
[0062]
8b(1
‑
(4
‑
甲氧基苄基)
‑5‑
甲基
‑
1,4
‑
二氢吡啶
‑3‑
甲酰胺):25%。
[0063][0064]
实施例3
[0065]
50mm 1ml ph 7.5tris
‑
hcl缓冲液中含有1mm二苯并噻吩、10mm nadh或mnadhs(1b
‑
8b)、10μm fmn、3u dszc酶、1u dszd酶、50u过氧化氢酶。37℃、200rpm,反应在密闭的1.5ml离心管中进行,分别取反应2h、4h、8h、12h、24h的反应液200μl,加入3
×
200μl乙酸乙酯萃取,超声混合、10000rpm离心2min,取有机相进行液相检测,转化率(%)如表1所示。
[0066]
表1
[0067]
时间(h)nadh1b2b3b4b5b6b7b8b257.2317.9320.7921.5322.1122.7815.0355.9859.87468.3621.6525.8139.8240.5138.9828.9984.7663.77899.2843.7850.7057.5866.3957.2239.7393.9282.101299.4176.9389.2396.2394.5881.1154.7897.9096.8824100.089.1492.5499.6998.6589.3267.8098.93100.0
[0068]
实施例4
[0069]
50mm 1ml ph 7.5tris
‑
hcl缓冲液中含有1mm二苯并噻吩、2
‑
30mm mnadhs(3b、4b、7b、8b)、10μm fmn、3u dszc酶、50u过氧化氢酶。37℃、200rpm,反应在密闭的1.5ml离心管中进行,12h后取各反应液200μl,加入3
×
200μl乙酸乙酯萃取,超声混合、10000rpm离心2min,取有机相进行液相检测,转化率(%)如表2所示。
[0070]
表2
[0071]
当量(mm)3b4b7b8b234.5544.1442.5547.93574.5579.1471.7877.93894.8497.5497.2190.691090.4394.6596.0096.931590.2892.5892.3391.92
2086.6390.1972.9083.162588.2189.2574.1284.093070.1969.2170.1165.45
[0072]
实施例5
[0073]
50mm 1ml ph 5
‑
9tris
‑
hcl缓冲液中含有1mm二苯并噻吩、10mm mnadhs(3b、4b、7b、8b)、10μm fmn、3u dszc酶、50u过氧化氢酶。37℃、200rpm,反应在密闭的1.5ml离心管中进行,12h后取各反应液200μl,加入3
×
200μl乙酸乙酯萃取,超声混合、10000rpm离心2min,取有机相进行液相检测,转化率(%)如表3所示。
[0074]
表3
[0075]
ph3b4b7b8b521.5323.1442.1133.21673.8570.5079.4777.40794.5398.3894.0099.76895.2597.5698.2197.90979.8580.5086.4081.44
[0076]
实施例6
[0077]
50mm 1ml ph 7.5tris
‑
hcl缓冲液、1mm二苯并噻吩、10mm mnadhs(3b、4b、7b、8b)、10μm fmn、3u dszc酶、50u过氧化氢酶。25
‑
40℃、200rpm,反应在密闭的1.5ml离心管中进行,12h后取各反应液200μl,加入3
×
200μl乙酸乙酯萃取,超声混合、10000rpm离心2min,取有机相进行液相检测,转化率(%)如表4所示。
[0078]
表4
[0079]
温度℃3b4b7b8b2580.1083.9284.3481.343090.2592.3298.2897.983592.1393.0191.6294.204051.2259.5661.7860.22
[0080]
实施例7
[0081]
50mm 1ml ph 7.5tris
‑
hcl缓冲液、1mm二苯并噻吩、8mm mnadhs、10μm fmn、二苯并噻吩单加氧酶、50u过氧化氢酶。30℃、200rpm,反应在密闭的1.5ml离心管中进行,2
‑
12h后取各反应液200μl,加入3
×
200μl乙酸乙酯萃取,超声混合、10000rpm离心2min,取有机相进行液相检测,转化率(%)如表5所示。
[0082]
表5
[0083]
时间nadh
a
1b
b
3b
b
4b
b
7b
b
8b
b
2h58.1148.1253.2151.8954.6261.804h75.5265.9882.2581.7292.2888.988h98.9089.0296.3395.9697.6295.2012h10097.45100100100100
[0084]
备注:a:二苯并噻吩单加氧酶为3u dszc酶、1u dszd酶;b:二苯并噻吩单加氧酶为
3u dszc酶。
[0085]
本发明提供了一种人工烟酰胺辅因子介导的酶法降解二苯并噻吩的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。