1.本发明涉及稳定且生物利用度高而给药效果优异的艾氟康唑口服用组合物。
背景技术:
2.艾氟康唑是在甲真菌病的治疗中得到活性认证的三唑类化合物,其抑制真菌细胞膜的主成分-麦角甾醇的合成,表现出抗真菌作用。
3.目前商业化产品有朱布利亚(jublia)
tm
,朱布利亚采用艾氟康唑作为主要成分,用于皮肤癣菌引起的甲真菌病的治疗剂,在2017年5月获得食品医药品安全部门的产品许可,并正式在韩国推出。
4.朱布利亚为局部涂抹剂型,局部抗真菌剂没有皮疹、肝毒性和消化系统副作用等口服抗真菌剂的缺点,有着高药物依从性和偏好度。
5.甲真菌病的治疗中,可用于艾氟康唑及其他三唑类抗真菌药物的局部传递的剂型示例在如美国专利号8486978号(专利文献1)等中。
6.但是,局部涂抹剂型的情况下,由于具有穿透外皮或指甲等的特性,在抑制根深蒂固的真菌方面有些不足,并且,因需要涂抹于所有病变部位的特点,对于广泛扩散于全身等的真菌的抑制和预防有所受限
7.事实上,真菌性疾病,例如手部和足部或者手指甲和脚指甲上的癣容易再发的原因与所述局部涂抹剂型的局限性息息相关。即,虽然局部涂抹剂型能够很好地作用于表面常在的真菌,但在所需的有效浓度下,无法作用到真菌能够被完全杀灭的深度。
8.因此,将抗真菌成分通过血液等的途径传递到相应组织的给药方法(例如,注射剂、口服用片剂、液剂、胶囊剂、栓剂等)的开发较为重要,尤其是,需要开发方便食用且服药依从性和偏好度高的口服型剂型。
9.但是,艾氟康唑的水溶解度为较低的0.61mg/ml,口服给药时可能在吸收率和生物利用度上产生问题。
10.并且,含有艾氟康唑的溶液制剂有着稳定性问题,即,在短储存期内发生变色,有着组合物的颜色从黄色变为深红色或褐色范围的问题。为了解决上述稳定性问题,美国专利号us9662394号公开了包含特定螯合剂、抗氧化剂、以及酸,即,乙二胺四乙酸(edta)或其盐、丁基羟基甲苯(bht)、以及柠檬酸的液体或半固体组合物。
11.因此,需要开发生物利用度高且稳定的艾氟康唑口服用制剂。
12.作为参考,可引用并综合下述先行技术文献全部内容作为本发明的背景技术内容。
13.(专利文献1)us8486978b2(2013.07.16)
14.(专利文献2)us9662394b2(2017.05.30)
15.(专利文献3)us5620994a(1997.04.15)
技术实现要素:
16.本发明为了解决上述现有技术问题,提供生物利用度高且稳定的艾氟康唑口服用制剂。
17.本发明为解决上述现有技术的问题而提出的,
18.提供一种口服用药物组合物,其特征在于,包含艾氟康唑、聚乙二醇的共形成产物和药学上可接受的添加剂。
19.另外,本发明提供一种口服用药物组合物,其特征在于,所述组合物为片剂或液剂。
20.此外,本发明提供一种口服用药物组合物,其特征在于,包含0.6-2040mg的艾氟康唑。
21.另外,本发明提供一种口服用药物组合物,其特征在于,所述共形成产物中,艾氟康唑与聚乙二醇的重量比为1:1。
22.此外,本发明提供一种口服用药物组合物,其特征在于,所述药学上可接受的添加剂为选自赋形剂、粘合剂、崩解剂、表面活性剂、润滑剂和着色剂中的一种以上。
23.另外,本发明提供一种口服用药物组合物,其特征在于,所述赋形剂为选自乳糖水合物、微晶纤维素、微晶纤维素-乳糖、甲基纤维素、乙基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素盐、其他取代和未取代的纤维素、玉米淀粉、糊化淀粉、乳糖、无水乳糖、糖醇、蔗糖、木糖醇、丙烯酸酯聚合物和共聚物、葡萄糖结合剂、糊精、葡萄糖、麦芽糊精、果胶、明胶和无机稀释剂中的一种以上。
24.此外,本发明提供一种口服用药物组合物,其特征在于,所述粘合剂选自聚乙烯醇-聚乙二醇接枝共聚物、聚乙烯吡咯烷酮乙酸乙烯酯、乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素、甲基纤维素、聚乙烯醇、聚乙烯吡咯烷酮、聚丙烯酸、明胶、丙二醇、海藻酸钠和它们的混合物。
25.另外,本发明提供一种口服用药物组合物,其特征在于,所述崩解剂为选自交联羧甲基纤维素钠、交联聚维酮、羧甲基纤维素盐、羟丙基纤维素、微晶纤维素、糊化淀粉、淀粉钠、乙醇酸盐和淀粉乙醇酸钠中的一种以上。
26.此外,本发明提供一种口服用药物组合物,其特征在于,所述润滑剂选自硬脂酸镁、硬脂富马酸钠和山嵛酸甘油酯。
27.另外,本发明提供一种口服用药物组合物,其特征在于,其用途为预防或治疗真菌感染症。
28.此外,本发明提供一种口服用药物组合物,其特征在于,所述真菌感染症为甲真菌病。
29.发明效果
30.本发明体现出高稳定性和生物利用度,有着适用于口服用药的效果。
附图说明
31.图1是关于艾氟康唑和聚乙二醇共晶的粉末xrd图。
32.图2是关于艾氟康唑和聚乙二醇共晶的dsc热分析图。
33.图3是关于艾氟康唑结晶型的粉末xrd图。
34.图4是关于艾氟康唑结晶型的dsc热分析图。
35.图5是艾氟康唑结晶型和艾氟康唑聚乙二醇共晶(co-crystal)的稳定性的比较图。
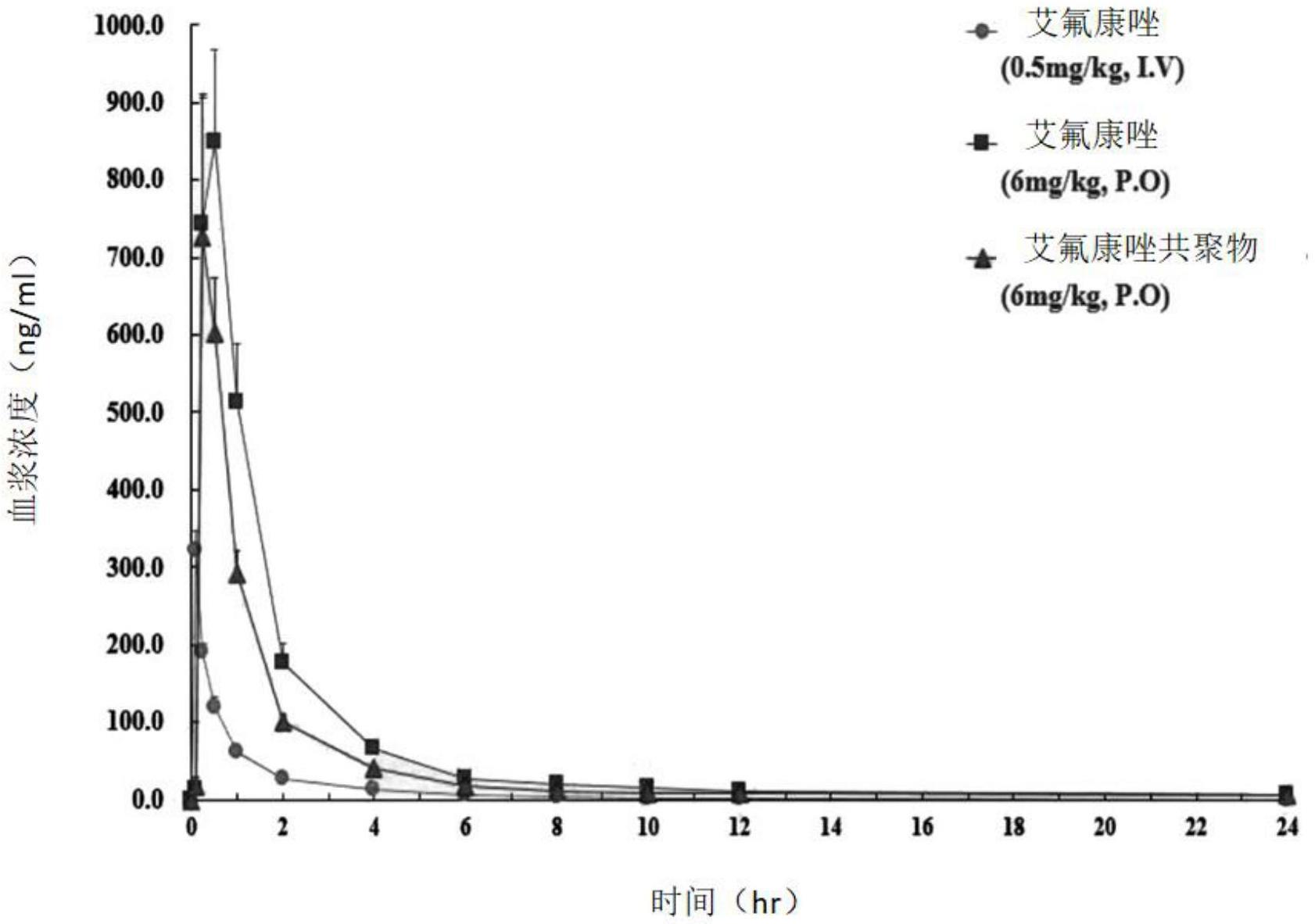
36.图6是显示临床前实验中伏立康唑的时间-血液浓度的曲线图。
37.图7是显示临床前实验中艾氟康唑的时间-血液浓度的曲线图。
具体实施方式
38.下面详细说明本发明。
39.本发明涉及口服用药物组合物,其中,包含艾氟康唑、聚乙二醇的共形成产物和药学上可接受的添加剂。
40.共形成产物是两种以上物质表现出像一种单独的物质一样独特(unique)的物理特性的物质,可分类为非晶形态的共非晶(co-amorphous)和结晶型形态的共晶(co-crystal)。
41.所述共晶是指两种以上的不同分子在一个晶格中以特定的化学计量比(stoichiometric ratio)形成结晶结构的形态,共晶内分子间键的形态有别于盐类和混合物。
42.此外,共晶(cocrystal)是指具有丰富的能形成氢键的官能团如o、oh、n等或通过共形成分子(coformer)与氢键按常规比例结合而具有新的结晶结构的结晶性固体。
43.所述共形成剂(coformer)是指构成共晶医药品结晶的分子中没有活性的分子,本发明中优选为聚乙二醇。
44.本发明涉及的关于艾氟康唑和聚乙二醇共晶的粉末xrd图和dsc热分析图如图1和图2所示。关于艾氟康唑结晶型的xrd图和dsc热分析图如图3和图4所示。
45.比较图1至图4,可以确认本发明涉及的艾氟康唑共形成产物相对于艾氟康唑结晶型具有完全不同的趋势。
46.也就是说,艾氟康唑聚乙二醇的共形成产物具有如下结晶结构,即,在粉末xrd图特征性地显示7.78、11.50、13.85、15.49、16.79、18.97、19.22、23.57、26.18、27.09的衍射角(2θi/i0》10%),通过差示扫描量热法(dsc)分析的光谱上,在61.62℃和78.78℃处观察到最大吸热峰,上述xrd图或dsc热分析图相对于艾氟康唑结晶型是明显不同的。
47.因此,可以确定本发明的艾氟康唑的共形成产物是一种理化性质与现有艾氟康唑结晶型完全不同的新型物质。
48.并且,由于艾氟康唑是不稳定状态的药物,为了作为药物组合物的活性成分使用,需要将其稳定性提高,通过形成共形成产物,可以提高其稳定性。
49.即,如图5所示,本发明的艾氟康唑的共形成产物的稳定性优异。
50.而且,艾氟康唑的共形成产物可以增加药物溶解度及溶解速率,改变人体内对药物的吸收率,从而改变生物利用度。
51.即,如图6所示,本发明涉及的艾氟康唑共形成产物的生物利用度非常优异。
52.以上述特征为基础,本发明涉及的艾氟康唑的共形成产物能够全部满足作为药物组合物的活性成分所需的要求,即结晶的化学性纯度、组合物溶液、温度的稳定性和口服给药时的生物利用度等。
53.关于本发明涉及的艾氟康唑的共形成产物的稳定性或生物利用度通过后述的实施例进行详细地说明。
54.本发明的口服用药物组合物中,作为有效成分的艾氟康唑,根据稳定性和有效性资料,包含预防或者治疗有效量。在部分具体例中,治疗有效量为约0.01至约30mg/kg;部分特定的具体例中,可以为约0.01至约6mg/kg。因此,口服用给药剂型的艾氟康唑含量为0.6-2040mg,或包含0.6-410mg,但不一定限定于此,根据临床发现可进行变动。
55.所述药学上可接受的添加剂可以非限制性地使用对本领域技术人员而言显而易见且熟知的药学添加剂,其实例为稳定剂、表面活性剂、润滑剂、增溶剂、缓冲剂、甜味剂、基剂、吸附剂、矫味剂、粘合剂、悬浮剂、固化剂、抗氧化剂,增亮剂、香料、调味剂、颜料、涂料剂、润湿剂、润湿控制剂、填料、消泡剂、制冷剂、咀嚼剂、防静电剂、着色剂、糖衣剂、等渗剂、柔顺剂、乳化剂、粘附剂、增稠剂、发泡剂、ph调节剂、赋形剂、分散剂、崩解剂、防水剂,防腐剂、保鲜剂、溶解助剂、溶剂和流动化剂等;更具体地,药学上可接受的添加剂可以选自赋形剂、粘合剂、崩解剂、表面活性剂、润滑剂和着色剂中的一种以上。
56.所述赋形剂实例为选自乳糖水合物、微晶纤维素、微晶纤维素-乳糖、甲基纤维素、乙基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素盐、其他取代和未取代的纤维素、玉米淀粉、糊化淀粉、乳糖、无水乳糖、糖醇、蔗糖、木糖醇、丙烯酸酯聚合物和共聚物、葡萄糖结合剂、葡聚糖、葡萄糖、麦芽糊精、果胶、明胶和无机稀释剂中的一种以上,
57.所述粘合剂实例为选自聚乙烯醇-聚乙二醇接枝共聚物、聚乙烯吡咯烷酮乙酸乙烯酯、乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素、甲基纤维素、聚乙烯醇、聚乙烯吡咯烷酮、聚丙烯酸、明胶、丙二醇、海藻酸钠和它们的混合物,
58.所述崩解剂实例为选自交联羧甲基纤维素钠、交联聚维酮、羧甲基纤维素盐、羟丙基纤维素、微晶纤维素、糊化淀粉、淀粉钠、乙醇酸盐和淀粉乙醇酸钠中的一种以上,
59.所述表面活性剂实例为十二烷基磺酸钠(sodium dodecyl sulfonate,sds)、十二烷基硫酸钠(sodium lauryl sulfate,sls)、嵌段共聚物(pluronic)f127、嵌段共聚物(pluronic)f68、聚(乙二醇)(poly(ethylene glycol),peg)、聚氧乙烯山梨醇单油酸酯(polyoxyethylene sorbitan monooleate,tween 80)、泊洛沙姆(poloxamer,lutrol f68)、聚乙二醇-15-羟基硬脂酸酯(polyethylene glycol-15-hydroxystearate,solutol hs15)、羟丙基甲基纤维素(hydroxypropyl methyl cellulose,hpmc)、聚(n-乙烯基-2-吡咯烷酮)(poly(n-vinyl-2-pyrrolidone),pvp)、silwet l77、聚乙烯醇(poly(vinylalcohol),pva),牛血清白蛋白(bovine serum albumin,bsa)和蔗糖脂肪酸酯(sucrose fatty acid esters)等,
60.所述润滑剂实例选自硬脂酸镁、硬脂富马酸钠和山嵛酸甘油酯,但并不限定于此,可以非限制性地使用对本领域技术人员而言显而易见且熟知的药学添加剂。
61.本发明的组合物,可用于真菌感染症的预防或治疗。适应症的具体示例包括但不限于念珠菌性阴道炎、花斑癣、由皮肤癣菌引起的体部白癣、股部白癣(股癣)、手部白癣、足部白癣、口腔念珠菌感染、真菌性角膜炎、甲真菌病、曲霉病、念珠菌感染、念珠菌尿、念珠菌血症、隐球菌病(包括隐球菌脑膜炎),巴西芽生菌病、系统性真菌感染、脂溢性皮炎和酵母引起的头皮屑等。
62.以下,通过参照实施例更具体地解释本发明。然而,要注意的是,这些实施例只是为了更详细地说明发明,并不是限制本发明的范围。
63.实施例
64.实施例1-艾氟康唑和聚乙二醇的共晶制备(1:1比例)
65.将艾氟康唑(1g)在常温下搅拌溶解在乙腈(5ml)中。向该反应液中加入聚乙二醇-6000(1g),搅拌1小时后,减压蒸馏除去溶剂,短时间内自发形成艾氟康唑和聚乙二醇-6000共晶的结晶型。真空干燥下仅回收结晶,收率为95%,纯度为99.5%。所述实施例1中制备的艾氟康唑和聚乙二醇-6000共晶的粉末xrd光谱(x-ray diffraction spectrum)和差示扫描量热仪热谱图(differential scanning calorimeter thermogram)分别如图1和图2所示。
66.实施例2-口服用液剂的制备
67.将实施例1中制备完成的产物和将0.5%甲基纤维素分散在含有表面活性剂1%的蒸馏水中的溶液进行涡流(vortex)并超声处理,以制备均匀的口服用液剂。具体的成分配比如下述表1所示。
68.【表1】
[0069][0070]
实施例3-口服用片剂的制备
[0071]
将实施例1中制备完成的产物和赋形剂捏合,并依次向其中加入崩解剂和润滑剂,将粒子进行整粒后,进行压片得到未包衣的片剂,具体的成分配比如下述表2所示。
[0072]
【表2】
[0073][0074]
实施例4-口服用胶囊剂的制备
[0075]
将实施例3中制备的制剂不进行压片,放入明胶胶囊制备胶囊剂。
[0076]
测试例
[0077]
【稳定性测试】
[0078]
为了确认关于本发明的艾氟康唑共晶对温度和组合物溶液的稳定性,进行了以下
实验来比较艾氟康唑对温度和组合物溶液的稳定性。
[0079]
*实验方法
[0080]
在常温和65℃烘箱中,将艾氟康唑结晶型和实施例1以组合物溶液中溶解的状态分别放置一周、两周和四周,然后通过高效液相色谱(hplc)进行分析来测定纯度。
[0081]
其结果如下述表3和图5所示。
[0082]
【表3】
[0083][0084]
从表3和图5可以看出,与现有的艾氟康唑结晶型相比,本发明特征的艾氟康唑共晶在温度和溶解于组合物溶液的状态中更稳定,在药物制备上具有更多有用的优点。而且,本发明的艾氟康唑共晶为新物质,均满足药物组合物的活性成分的所有要求。
[0085]
【临床前试验】
[0086]
将24只雄性比格犬分为4组,每组6只,在禁食条件下,将对照药伏立康唑通过口服途径给动物服用单次,将试验药艾氟康唑通过静脉和口服途径给动物服用单次,之后获得各药物的pk profile,以计算药物的pk参数(parameter)并评估药代动力学。具体试验条件和方法如下。
[0087]
1、试验药的制备
[0088]
试验药为口服给药组的情况下,在进行试验药物制备时,根据测量的体重,用电子天秤称量基于给药用量测量的伏立康唑(口服给药g1组)、艾氟康唑(口服给药g2组)、所述实施例1中制备的艾氟康唑共晶(口服给药g3组-efinaconazole copolymer)后,加入到玻璃容器中,使用赋形剂(1%tween 80溶液(distilled water)中的0.5%甲基纤维素),进行涡流(vortex)和超声波处理,使其制备成均匀的状态。
[0089]
静脉给药的情况下,加入基于给药用量测量的艾氟康唑(静脉给药g1组)和作为赋形剂的相当于总配方体积的30%的peg400后,通过涡流(vortex)和超声波使其完全溶解,加入相当于总配方体积的20%的乙醇后,通过超声波进行均质化,然后,加入相当于总配方体积的50%的0.9%生理盐水(saline),之后通过涡流(vortex)和超声波处理,制备均质状态的试验药。
[0090]
2、给药
[0091]
–
给药途径:静脉给药(iv),口服给药(po)
[0092]
–
给药用量:g1【伏立康唑(voriconazole)】-6mg/5ml/kg(po);g2【艾氟康唑(efinaconazole)】-6mg/5ml/kg(po);g3【艾氟康唑聚合物(efinaconazole-polymer)】-6mg/5ml/kg(po);g1【艾氟康唑(efinaconazole)】-0.5mg/1ml/kg(iv)
[0093]
–
给药频率:单次(1日1次)-(qd)
[0094]
依据给药的试验组构成如下述表4所示。
[0095]
【表4】
[0096]
·
口服给药
[0097][0098]
·
静脉给药
[0099][0100]
3、试验结果
[0101]
进行试验时,分配方式为口服给药g1组为1-6号,g2组为7-12号,g3组为13-18号,静脉给药g1组为1-6号。试验当天,确认各组的平均体重为口服给药g1组为11.5
±
1.2kg,g2组为11.5
±
1.3kg,g3组为11.5
±
1.0kg,静脉给药g1组为11.6
±
1.6kg,
[0102]
在试验过程中,未观察到与试验物质给药相关的异常症状(包括呕吐、跛行、腹泻和行为异常等),在整个试验期间,所有个体都没有发生个体死亡病例,采血工作进展顺利。每个点收集的采血量为4ml,将采集的血液进行离心分离后,将500μl的血浆上清液分注到1.5ml的ep tube中,将剩余的血浆分注到其余的1.5ml的ep tube 1set中,储存在-80℃的冷冻箱(deep freezer)中,试验结束后,将血液采集记录本、样本信息证明书、配药记录书、临床症状观察记录书、用药/血液采集结果和试验样本(血浆)递送给专业分析机构。
[0103]
递送的样本在进行分析为止保管在冷冻箱中,通过lc/ms/ms对样本进行分析,使用“phoenix winnonlin(version 8.2or higher certara l.p)”用noncompartmental方法计算药代动力学参数。
[0104]
将血浆样本进行分析时,伏立康唑(6mg/kg,p.o)组分配为g1组(no.1-6),艾氟康唑组分别分配为g2组-0.6mg/kg i.v(no.1-6),g3组-6mg/kg p.o(no.7-12),g4组-共聚物6mg/kg p.o(no.13-18)进行分析,如下述表5所示。
[0105]
【表5】
[0106][0107]
每组分配6只,进行单次给药后,将总数24只的试验试料进行分析的伏立康唑和艾氟康唑的血液中浓度趋势如图6和7以及表6和7所示。检测到峰值的试料中,低于最低定量限的试验试料使用blq进行处理。
[0108]
【表6】
[0109]
单位:mg/ml
[0110][0111]
【表7】
[0112]
单位:mg/ml
[0113][0114]
auct为生物利用度参数,是从给药时间到可测量的最后一次采血时间点的血浆浓度-时间曲线下的面积,通过梯形公式计算。c
max
是从每个个体的血浆药物浓度-时间曲线读取的最高血浆药物浓度,t
max
是从每个个体的血浆药物浓度-时间曲线中达到最高血浆药物浓度所花费的时间。auc
∞
是从给药时间到无穷时间的血浆浓度-时间曲线下的面积。对伏立康唑和艾氟康唑计算的pk参数如下述表8所示。
[0115]
【表8】
[0116][0117]
如上述表所示,pk参数中伏立康唑(6mg/kg,p.o)组、艾氟康唑(0.6mg/kg,i.v)组、艾氟康唑(6mg/kg,p.o)组、艾氟康唑共聚物(6mg/kg,p.o)组的auc
t
分别显示为53905.19
±
7138.87、253.28
±
74.20、1494.88
±
475.14和987.98
±
271.62;auc
∞
值确认为109208.24
±
24516.63、270.71
±
84.91、1610.32
±
540.33和1114.49
±
323.58;c
max
值确认为3674.45
±
169.60、322.20
±
58.61、925.10
±
236.42和827.06
±
349.93。
[0118]
在统计分析结果中,与艾氟康唑(6mg/kg,p.o)组相比,试验药艾氟康唑共聚物(6mg/kg,p.o)组的c
max
和auc
t
指标表现出显著较低的结果。(下述表9至表12)【表9】
[0119][0120]
【表10】
[0121][0122]
【表11】
[0123][0124]
【表12】
[0125][0126]
艾氟康唑共聚物(6mg/kg,p.o)组和艾氟康唑(6mg/kg,p.o)组的生物利用度分别为35.69
±
22.61%和51.36
±
24.63%(下述表13和表14)。
[0127]
【表13】
[0128][0129]
f(%)=[auc(p.o.)/dose(p.o.)]/[auc(i.v.)/dose(i.v.)]*100
[0130]
【表14】
[0131][0132]
f(%)=[auc(p.o.)/dose(p.o.)]/[auc(i.v.)/dose(i.v.)]*100结果上,与艾氟康唑(6mg/kg,p.o)组相比,艾氟康唑共聚物(6mg/kg,p.o)组的auc
t
数值相当于66%水平,显著较低,但考虑到共聚物剂型中原料成分的含量为50%,因此可以确认超过了此水平。
[0133]
并且,艾氟康唑共聚物(6mg/kg,p.o)组的生物利用度为35.69%,在用量线型性成
立的条件下,与显示51.36%的艾氟康唑(6mg/kg,p.o)组进行比较得出约达到其70%的水平,表明试验物质艾氟康唑共聚物剂型在比格犬体内的暴露度远高于理论预期水平(50%)。
[0134]
从这些测试结果得出,即使在低剂量口服时,艾氟康唑的生物利用度也高于其他抗真菌药物,例如伏立康唑和伊曲康唑等,因此,通过口服给药可以期待其较高治疗效果。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。