原位atrp引发的有机/无机杂化阴离子交换膜及其制备方法
技术领域
1.本发明属于新材料和高分子材料领域,具体涉及一种原位atrp引发的有机/无机杂化阴离子交换膜及其制备方法。
背景技术:
2.目前,碱性电解水制氢技术已经引起了广泛关注,其可以利用从风能和太阳能等可再生能源中获得的电能,将水分成高纯度的氢气和氧气,是氢能社会的重要技术之一。其中,碱性阴离子交换膜电解水制氢技术采用高分子聚合物作为固态电解质,结合了质子交换膜电解水制氢和液体koh电解水制氢技术的优点,具有突出优势。在碱性条件下,廉价的不含铂族金属的催化剂可以用于阴离子交换膜电解水制氢,此外,当用固体阴离子交换膜代替液体碱性电解质(35wt%koh)时,可以降低腐蚀性,同时提升电解槽对co2和压差的耐受性。
3.然而,阴离子交换膜电解水制氢技术的发展仍处于早期阶段,其性能和耐久性受限于核心部件
‑‑
阴离子交换膜(aem)的离子导电性和稳定性。增加aem的离子交换容量(iec)可以提升阴离子交换膜的电导率,有效地降低电解槽的欧姆电阻,从而改善aem电解槽的性能。目前,离子交换容量、分子量的准确控制可以通过活性原子转移自由基聚合(atrp)来实现,该方法可精确控制侧链上离子交换基团的数量和分布,以及侧链长度,以平衡稳定性和导电性。zhu等人通过atrp的方法制备了交联的萘基嵌段阴离子交换膜,具有高碱性稳定性、低膨胀率和高离子传导性(zhu z y,gou w w,chen j h,et al.journal of membrane science,2021,636:119569.)。通过原子转移自由基聚合的手段已被广泛用于合成具有可控结构的共聚物。
4.虽然通过引入较多的阳离子基团来提高离子交换容量,可以促进离子电导率,但也可能会导致膜材料高度水合和溶胀,进而丧失膜的化学和尺寸稳定性。一方面,可以通过化学交联限制分子链的移动来有效地提高aem的尺寸稳定性,特别是那些具有较高iec的聚合物分子;另一方面,无机掺杂如引入零维、一维和二维等不同尺度的无机填料,也可以增加膜的尺寸稳定性。目前,氧化石墨烯、金属有机框架(mofs)、和层状双氢氧化物(ldhs)等已被报道在复合aem中具有增强抗溶胀性和导电性的作用。专利申请cn104231294b公布了一种氧化石墨烯复合的聚苯醚作为有机组份,制备纳米复合阴离子交换膜,其加入可以大幅度提高阴离子交换膜的机械性能。而如何制备可靠的、有机/无机相容性高的复合阴离子交换膜仍是一项挑战。
技术实现要素:
5.为解决上述问题,本发明公开一种原位atrp引发的有机/无机杂化阴离子交换膜及其制备。
6.本发明通过原子转移自由基聚合,精确控制阴离子交换膜的离子交换容量(1.85mmol/g干膜)和分子量(mw=9320),并在此基础上,通过无机掺杂提高阴离子交换膜
的尺寸稳定性、热稳定性、和机械性能,促进离子交换膜综合性能提升,通过溴化的无机填料原位引发高分子单体聚合,提升有机/无机杂化的界面相容性。我们在此报告了由碳纳米管负载的有机/无机杂化阴离子交换膜,表现出较高离子交换容量,并在80℃时具有高氢氧化物传导率。此外,此膜还呈现出明显的高碱性稳定性和机械性能,赋予相应的电解水制氢和燃料电池提升的功率密度和良好的耐久性。该功能化高性能复合膜将为碱性电解水制氢和燃料电池关键材料的结构设计开辟新途径,推动氢能技术的可靠应用和持续发展。
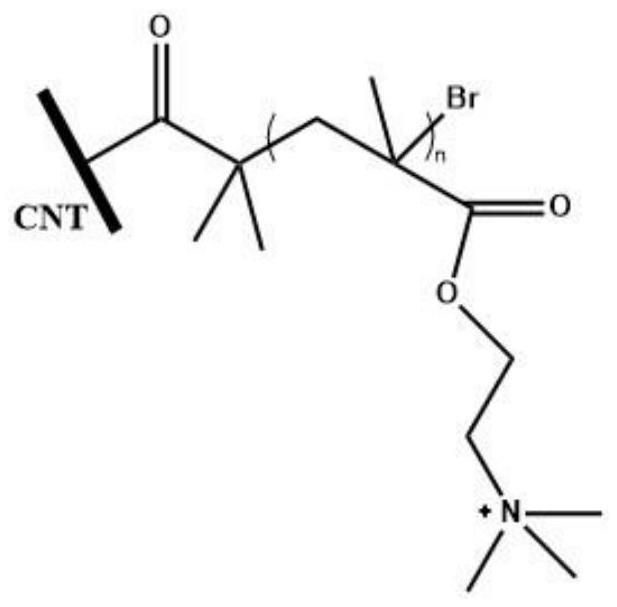
7.本发明提出的一种原位atrp引发的有机/无机杂化阴离子交换膜,其特征在于以溴化的碳纳米管(cnt)同时作为引发剂和无机填料,引发单体n,n-甲基丙烯酸二乙胺基乙酯聚合,得到高分子骨架,分子侧链末端经与甲基化试剂的门秀特金反应,得到季铵离子作阴离子交换位点,其结构式如下所示,
[0008][0009]
本发明提出的一种原位atrp引发的有机/无机杂化阴离子交换膜的制备方法,具体步骤如下:
[0010]
(1)将羟基化碳纳米管分散在溶剂a中,混合物被超声处理10~60分钟,然后,加入硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流12~48小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在40~60℃下真空干燥;
[0011]
(2)将br@cnts、单体n,n-甲基丙烯酸二乙胺基乙酯溶解在溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入催化剂b和配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在60~120℃下反应24~48小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂c,沉淀得到产物并真空干燥;
[0012]
(3)将步骤(2)得到的产物溶解在四氢呋喃中,加入甲基化试剂d,在40~60℃下反应24~72小时,用沉淀剂c沉淀洗涤得到产物并真空干燥;
[0013]
(4)将步骤(3)得到的产物溶解在溶剂e中,配制膜溶液浓度为5wt%~20wt%,过滤后铸膜,在50~70℃下烘干5~24h,得到阴离子交换膜。
[0014]
本发明中,所述溶剂a为甲苯、甲醇、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、n,n-二乙基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜中的一种或几种;所述催化剂b为溴化亚铜/联吡啶、氯化亚铜/联吡啶、溴化亚铜/五甲基二乙烯基三胺、氯化亚铜/五甲基二乙烯基三胺、溴化亚铜/三(2-甲氨基乙基)胺、氯化亚铜/三(2-甲氨基乙基)胺、溴化亚铜/六甲基三亚乙基四胺、氯化亚铜/六甲基三亚乙基四胺、溴化亚铜/2-吡啶甲醛缩正丙胺或氯化亚铜/2-吡啶甲醛缩正丙胺中的一种或几种;所述沉淀剂c为乙酸乙酯、乙醚、二甲苯、环己烷或正己烷中的一种或几种;所述甲基化试剂为碘甲烷、溴甲烷、三氟碘甲烷、三氟碘乙烷、碘
乙烷、1-氟-2-碘乙烷;所述溶剂e为二甲基亚砜、二甲基甲酰胺、甲基吡咯烷酮、二甲基乙酰胺中的一种或几种;所述碳纳米管的质量百分比为5wt%~20wt%。
[0015]
与现有技术相比,本发明的有益效果在于:本发明提供的制备方法,其原料来源广泛,所用的碳纳米管、n,n-甲基丙烯酸二乙胺基乙酯、甲基化试剂、催化剂、溶剂等均可工业化生产,合成方法简单易行。合成的一种原子转移自由基聚合的有机/无机杂化阴离子交换膜具有精确控制的离子交换容量,且在无机掺杂物的支撑下,尺寸稳定且抗溶胀,热性能优异,实现可靠阴离子交换膜的制备。所得的原位原子转移自由基聚合引发的有机/无机杂化阴离子交换膜在碱性电解水制氢、碱性燃料电池、阴离子渗析等领域将具有广泛应用。
附图说明:
[0016]
图1:实施例1制备的一种原位atrp引发的有机/无机杂化阴离子交换膜的结构示意图。
[0017]
图2:实施例1制备的一种原位atrp引发的有机/无机杂化阴离子交换膜的离子电导率-温度柱状图。
[0018]
图3:实施例1制备的一种原位atrp引发的有机/无机杂化阴离子交换膜的凝胶渗透色谱(gpc)结果图。
具体实施方式
[0019]
以下实施例是对本发明的进一步说明,而不是限制本发明的范围。
[0020]
实施例1:
[0021]
(1)称取1.0g羟基化碳纳米管分散在20ml溶剂甲苯中,混合物被超声处理10分钟,然后,加入2ml硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流12小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在40℃下真空干燥;
[0022]
(2)称取0.5g br@cnts、2.0g单体n,n-甲基丙烯酸二乙胺基乙酯溶解在20ml溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入0.05g催化剂溴化亚铜/联吡啶和0.62g配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在60℃下反应24小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂乙醚,沉淀得到产物并真空干燥;
[0023]
(3)将步骤(2)得到的产物溶解在10ml四氢呋喃中,加入10ml甲基化试剂碘甲烷,在40℃下反应24小时,用沉淀剂乙醚沉淀洗涤得到产物并真空干燥;
[0024]
(4)称取步骤(3)得到的产物溶解在溶剂二甲基亚砜中,配制膜溶液浓度为5wt%,过滤后铸膜,在50℃下烘干5h,得到阴离子交换膜。最终得到的一种原位atrp引发的有机/无机杂化阴离子交换膜的结构式如图1所示,其离子电导率-温度关系如图2所示。
[0025]
上述技术方案,通过原子转移自由基聚合,可控合成该膜的分子量和离子交换容量;通过无机物复合,提升膜的抗溶胀性和尺寸稳定性;由该无机物原位引发后续单体进行原子自由基聚合,可增加有机/无机复合膜的相容性和可靠性。
[0026]
具体的,本发明通过原子转移自由基聚合,精确控制阴离子交换膜的离子交换容量(1.85mmol/g干膜,测试方法为酸碱滴定法)和分子量(mw=9320,见附图3),并在此基础
上,通过无机掺杂提高阴离子交换膜的尺寸稳定性、热稳定性、和机械性能,促进离子交换膜综合性能提升,通过溴化的无机填料原位引发高分子单体聚合,提升有机/无机杂化的界面相容性。由碳纳米管负载的有机/无机杂化阴离子交换膜,表现出较高离子交换容量,并在80℃时具有高氢氧化物传导率。此外,此膜还呈现出明显的高碱性稳定性和机械性能,赋予相应的电解水制氢和燃料电池提升的功率密度和良好的耐久性。
[0027]
实施例2:
[0028]
(1)称取1.2g羟基化碳纳米管分散在20ml溶剂甲醇中,混合物被超声处理20分钟,然后,加入2.5ml硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流16小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在45℃下真空干燥;
[0029]
(2)称取0.55g br@cnts、2.1g单体n,n-甲基丙烯酸二乙胺基乙酯溶解在20ml溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入0.06g催化剂氯化亚铜/联吡啶和0.64g配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在70℃下反应30小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂乙酸乙酯,沉淀得到产物并真空干燥;
[0030]
(3)将步骤(2)得到的产物溶解在10ml四氢呋喃中,加入10ml甲基化试剂碘乙烷,在45℃下反应30小时,用沉淀剂乙酸乙酯沉淀洗涤得到产物并真空干燥;
[0031]
(4)称取步骤(3)得到的产物溶解在溶剂二甲基亚砜中,配制膜溶液浓度为10wt%,过滤后铸膜,在55℃下烘干8h,得到阴离子交换膜。
[0032]
实施例3:
[0033]
(1)称取1.5g羟基化碳纳米管分散在20ml溶剂二氯甲烷中,混合物被超声处理30分钟,然后,加入2.8ml硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流20小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在50℃下真空干燥;
[0034]
(2)称取0.68g br@cnts、2.4g单体n,n-甲基丙烯酸二乙胺基乙酯溶解在20ml溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入0.07g催化剂溴化亚铜/联吡啶和0.72g配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在80℃下反应35小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂环己烷,沉淀得到产物并真空干燥;
[0035]
(3)将步骤(2)得到的产物溶解在15ml四氢呋喃中,加入15ml甲基化试剂溴甲烷,在50℃下反应35小时,用沉淀剂环己烷沉淀洗涤得到产物并真空干燥;
[0036]
(4)称取步骤(3)得到的产物溶解在溶剂二甲基亚砜中,配制膜溶液浓度为12wt%,过滤后铸膜,在60℃下烘干12h,得到阴离子交换膜。
[0037]
实施例4:
[0038]
(1)称取1.6g羟基化碳纳米管分散在20ml溶剂甲苯中,混合物被超声处理40分钟,然后,加入2.8ml硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流30小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在55℃下真空干
燥;
[0039]
(2)称取0.71g br@cnts、2.45g单体n,n-甲基丙烯酸二乙胺基乙酯溶解在20ml溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入0.08g催化剂溴化亚铜/联吡啶和0.83g配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在90℃下反应36小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂乙醚,沉淀得到产物并真空干燥;
[0040]
(3)将步骤(2)得到的产物溶解在15ml四氢呋喃中,加入15ml甲基化试剂碘甲烷,在55℃下反应48小时,用沉淀剂乙醚沉淀洗涤得到产物并真空干燥;
[0041]
(4)称取步骤(3)得到的产物溶解在溶剂二甲基亚砜中,配制膜溶液浓度为15wt%,过滤后铸膜,在65℃下烘干18h,得到阴离子交换膜。
[0042]
实施例5:
[0043]
(1)称取2.0g羟基化碳纳米管分散在20ml溶剂甲醇中,混合物被超声处理60分钟,然后,加入2ml硅烷偶联剂[4-(溴甲基)苯基]-三甲氧基硅烷,并在氮气环境下将悬浮液回流48小时,得到产物溴化的碳纳米管br@cnts,经离心并用乙醇洗涤,并在60℃下真空干燥;
[0044]
(2)称取0.75g br@cnts、2.5g单体n,n-甲基丙烯酸二乙胺基乙酯溶解在20ml溶剂无水二甲基甲酰胺中,充氮气搅拌均匀,向混合液中加入0.082g催化剂溴化亚铜/联吡啶和0.85g配位剂n,n,n',n,'n
”‑
五甲基二亚乙基三胺,并密封反应管,通过三次冷冻-泵-解冻循环对反应瓶进行脱气,并在120℃下反应48小时,终止反应后用四氢呋喃稀释混合物,通过中性氧化铝柱以除去催化剂,得到洗脱液,然后旋蒸浓缩,用沉淀剂乙酸乙酯,沉淀得到产物并真空干燥;
[0045]
(3)将步骤(2)得到的产物溶解在20ml四氢呋喃中,加入20ml甲基化试剂溴乙烷,在60℃下反应72小时,用沉淀剂乙酸乙酯沉淀洗涤得到产物并真空干燥;
[0046]
(4)称取步骤(3)得到的产物溶解在溶剂二甲基亚砜中,配制膜溶液浓度为20wt%,过滤后铸膜,在70℃下烘干24h,得到阴离子交换膜。
[0047]
上述描述仅是对本技术较佳实施例的描述,并非是对本技术范围的任何限定。任何熟悉该领域的普通技术人员根据上述揭示的技术内容做出的任何变更或修饰均应当视为等同的有效实施例,均属于本技术技术方案保护的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。