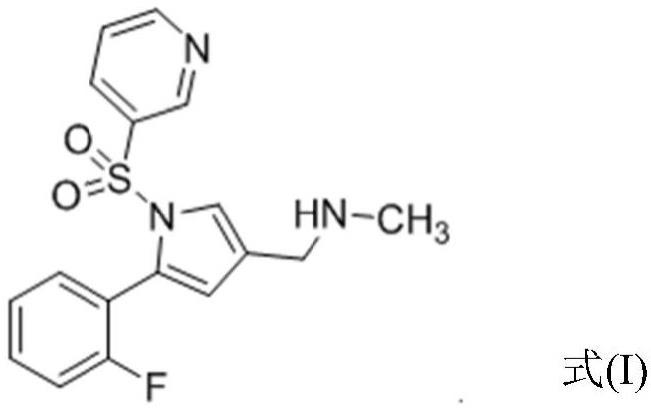
1.本发明涉及1-(5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基)-n-甲基甲胺(又名伏诺拉生)的固体形式,尤其涉及其氢溴酸盐晶型,制备所述固体形式的方法、包含所述固体形式的药物组合物,以及所述固体形式的用途。
背景技术:
2.伏诺拉生富马酸盐,又名tak438,是日本武田公司研制的一种钾离子竞争性酸阻滞剂(p-cab),其通过胃酸分泌抑制,发挥治疗胃酸相关疾病的作用。胃酸抑制作用最强的一类药物是质子泵抑制剂类(ppis),如奥美拉唑、兰索拉唑、雷贝拉唑等。相较于ppis类抑制剂,钾离子竞争性酸阻滞剂(p-cab)类药物,在临床上可明显减少夜间酸反跳的发生,疗效更稳定。
3.目前,已报道伏诺拉生的多种固体形式,其中,较多涉及富马酸盐及其晶型,例如富马酸盐三水合物、富马酸晶型i、富马酸晶型a。伏诺拉生富马酸盐水溶性差,公开文献显示富马酸伏诺拉生不同晶型的溶解度均小于1.0mg/ml;专利cn201710410953.9披露,即使溶解性较好的伏诺拉生二水合物晶型,其溶解性也仅达3.4-3.8mg/ml。富马酸盐的低溶解性,导致其口服生物利用度低,限制了其在抑酸和治疗胃酸相关疾病的应用。
4.针对上述问题,研究人员进一步研究伏诺拉生其他盐型及对应的晶型,例如醋酸盐及其晶型,但研究发现醋酸盐存在多种晶型,制备中极易产生混晶,不利于后续的放大生产。
5.因此,需要开发一种伏诺拉生的固体形式,以解决溶解性、稳定性、引湿性及工业化放大生产等问题,使其更利于药物加工和在药物组合物中使用,并为固体药物的疗效研究提供更多的定性定量信息,具有非常重要的意义,也是药物开发过程的迫切需求。
技术实现要素:
6.概述
7.本发明提供1-(5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基)-n-甲基甲胺(即式(i)化合物)的固体形式,
[0008][0009]
本发明一方面提供式(i)化合物的氢溴酸盐晶型i,所述式(i)化合物的氢溴酸盐
晶型i的x-射线粉末衍射(xrpd)图谱包括在13.8
±
0.2
°
、15.1
±
0.2
°
、18.3
±
0.2
°
、19.6
±
0.2
°
、21.9
±
0.2
°
、25.2
±
0.2
°
和25.9
±
0.2
°
处的衍射角(2θ)处的特征峰。
[0010]
本发明第二方面提供制备本发明式(i)化合物的氢溴酸盐晶型i的方法。
[0011]
本发明第三方面提供药物组合物,其包含本发明式(i)化合物的氢溴酸盐晶型i,以及一种或多种药学上可接受的载体。
[0012]
本发明第四方面提供本发明式(i)化合物的氢溴酸盐晶型i和/或本发明的药物组合物在制备用于预防和/或治疗糜烂性食管炎、胃溃疡、十二指肠溃疡、幽门螺杆菌根除及胃酸过多引起的相关疾病的药物中的用途。
[0013]
本发明第五方面提供预防和/或治疗糜烂性食管炎、胃溃疡、十二指肠溃疡、幽门螺杆菌根除及胃酸过多引起的相关疾病的方法,包括向需要其的个体给药有效量的本发明式(i)化合物的氢溴酸盐晶型i和/或本发明的药物组合物。
[0014]
发明详细描述
[0015]
定义
[0016]
除非在下文中另有定义,本文中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。提及本文中使用的技术意图指在本领域中通常所理解的技术,包括那些对本领域技术人员显而易见的技术的变化或等效技术的替换。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
[0017]
如本文中所使用的术语“包括”、“包含”、“具有”、“含有”或“涉及”及其在本文中的其它变体形式为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤,尽管其它未列举的元素或方法步骤不一定存在(即,这些术语也涵盖术语“基本上由
……
组成”和“由
……
组成”)。
[0018]
如本文中所使用的词语“约”是指本领域的普通技术人员认为在所述值的可接受的标准误差内,例如
±
0.05、
±
0.1、
±
0..2、
±
0.3、
±
0.5、
±
1、
±
2或
±
3等。。
[0019]
如本文中所使用的术语“固体形式”包括化合物i的所有固态形式,例如晶体形式。
[0020]
如本文中所使用的术语“晶型”或“晶体”是指呈现三维排序的任意固体物质,与无定形固体物质相反,其产生具有边界清楚的峰的特征性xrpd图谱。
[0021]
如本文中所使用的术语“x-射线粉末衍射图谱(xrpd图谱)”是指实验观察的衍射图或源于其的参数、数据或值。xrpd图谱通常由峰位(横坐标)和/或峰强度(纵坐标)表征。
[0022]
如本文中所使用的术语“2θ”是指基于x射线衍射实验中设置的以度数(
°
)表示的峰位,并且通常是在衍射图谱中的横坐标单位。如果入射束与某晶格面形成θ角时反射被衍射,则实验设置需要以2θ角记录反射束。应当理解,在本文中提到的特定晶体形式的特定2θ值意图表示使用本文所述的x射线衍射实验条件所测量的2θ值(以度数表示)。
[0023]
如本文中所使用的术语“差示扫描量热(dsc)图谱”是指由差示扫描量热仪记录到的曲线。
[0024]
如本文中所使用的术语“热重分析(tga)图谱”是指由热重分析仪记录到的曲线。
[0025]
如本文中所使用,术语“基本上相同”意指将代表性峰位和/或强度变化考虑在内。例如,对于x射线衍射峰,本领域技术人员会理解峰位(2θ)会显示一些变化,通常多达0.1-0.2度,并且用于测量衍射的仪器也会导致一些变化。另外,本领域技术人员会理解相对峰强度会因仪器间的差异以及结晶性程度、择优取向、制备的样品表面以及本领域技术人员
已知的其它因素而出现变化。
[0026]
晶型和制备方法
[0027]
第一方面,本发明提供式(i)化合物的氢溴酸盐晶型i,所述晶型i的x-射线粉末衍射(xrpd)图谱包括在13.8
±
0.2
°
、15.1
±
0.2
°
、18.3
±
0.2
°
、19.6
±
0.2
°
、21.9
±
0.2
°
、25.2
±
0.2
°
和25.9
±
0.2
°
衍射角(2θ)处的特征峰;
[0028][0029]
在部分实施方案中,本发明提供的式(i)化合物的氢溴酸盐晶型i中,式(i)化合物与氢溴酸的摩尔比为1:1。
[0030]
在部分实施方案中,所述式(i)化合物的氢溴酸盐晶型i的xrpd图谱包括在13.8
±
0.2
°
、15.1
±
0.2
°
、15.5
±
0.2
°
、18.3
±
0.2
°
、19.6
±
0.2
°
、21.9
±
0.2
°
、24.1
±
0.2
°
和24.3
±
0.2
°
、25.2
±
0.2
°
、25.9
±
0.2
°
和28.0
±
0.2
°
衍射角(2θ)处的特征峰。
[0031]
在部分优选实施方案中,所述式(i)化合物的氢溴酸盐晶型i的xrpd图谱包括在11.4
±
0.2
°
、13.8
±
0.2
°
、15.1
±
0.2
°
、15.5
±
0.2
°
、16.5
±
0.2
°
、18.3
±
0.2
°
、19.6
±
0.2
°
、20.8
±
0.2
°
、21.1
±
0.2
°
、21.9
±
0.2
°
、23.0
±
0.2
°
、24.1
±
0.2
°
、24.3
±
0.2
°
、25.2
±
0.2
°
、25.9
±
0.2
°
、26.4
±
0.2
°
、26.8
±
0.2
°
、27.8
±
0.2
°
、28.0
±
0.2
°
、29.5
±
0.2
°
、30.4
±
0.2
°
和31.3
±
0.2
°
衍射角(2θ)处的特征峰。
[0032]
在部分优选实施方案中,所述式(i)化合物的氢溴酸盐晶型i的xrpd图谱包括在以下衍射角(2θ)处的特征峰,其中2θ值的误差范围为
±
0.2
°
:
[0033]
2θ(
°
)
±
0.2
°
强度%2θ(
°
)
±
0.2
°
强度%9.60.4628.84.7911.410.5229.515.5713.879.9929.98.8815.138.8130.29.0615.525.4630.412.3715.95.7930.75.0216.514.9131.01.4718.334.5531.313.1818.65.5431.94.4519.610032.35.2720.59.1532.84.720.812.1433.49.4121.111.7733.67.8321.971.8434.71.8
22.24.5735.04.1323.018.8735.36.2224.123.6135.82.6724.322.8937.03.3225.235.937.29.6925.949.0137.74.8226.416.2137.81.5626.818.3838.51.4327.20.7238.71.3327.813.7639.41.2228.025.4339.84.53
[0034]
在更优选的实施方案中,所述式(i)化合物的氢溴酸盐晶型i包括与图1所示基本上相同的衍射角(2θ)处的峰。
[0035]
在最优选的实施方案中,所述式(i)化合物的氢溴酸盐晶型i的xrpd图谱如图1所示。
[0036]
在部分实施方案中,所述式(i)化合物的氢溴酸盐晶型i的差示扫描量热分析(dsc)图谱在201.5
±
5℃(起点温度)处出现吸热峰。
[0037]
在更优选的实施方案中,所述式(i)化合物的氢溴酸盐晶型i的dsc图谱包括如图2所示基本相同的温度处的特征峰。
[0038]
在更优选的实施方案中,所述式(i)化合物的氢溴酸盐晶型i的dsc图谱如图2所示。
[0039]
在部分实施方案中,所述式(i)化合物的氢溴酸盐晶型i的热重分析(tga)图谱在35℃~220℃无失重,温度高于220℃后开始分解。
[0040]
在部分优选的实施方案中,所述式(i)化合物的氢溴酸盐晶型i的tga图谱如图3所示。
[0041]
本发明的第二方面提供制备本发明式(i)化合物的氢溴酸盐晶型i的方法,所述方法包括以下步骤:
[0042]
步骤1:将式(i)化合物加入至第一溶剂中,升温至30-60℃,搅拌得到溶清液;
[0043]
步骤2:称量1.1-1.2倍(例如1.2倍)式(i)化合物摩尔比当量的氢溴酸,并加入第二溶剂稀释,得到酸溶剂;
[0044]
步骤3:将步骤2得到的酸溶剂,按一定滴加速度滴加至一定温度的步骤1的溶清液中;
[0045]
步骤4:降温至5-25℃并恒温养晶0.5-1h,过滤所得晶浆液,干燥至恒重,即得氢溴酸盐晶型i。
[0046]
在部分实施方案中,步骤1中所述第一溶剂选自甲醇、乙醇、丙酮、正丁醇、丁酮、环己酮、异丁醇、乙酸乙酯、乙酸异丙酯、1,4-二氧六环、乙腈、丁腈、仲丁醇、正丙醇、正戊醇和异戊醇中的一种或多种;
[0047]
优选地,步骤1中所述第一溶剂选自甲醇、丙酮和乙酸乙酯。
[0048]
在部分实施方案中,步骤1中所述溶清液中式(i)化合物的浓度为0.01-0.5g/ml,
优选为0.05-0.1g/ml;
[0049]
在部分实施方案中,步骤1中所述搅拌速度为200~500r/min,优选为250r/min。
[0050]
在部分实施方案中,步骤2中所述第二溶剂选自甲醇、乙醇、丙酮、正丁醇、丁酮、环己酮、异丁醇、乙酸乙酯、乙酸异丙酯、1,4-二氧六环、乙腈、丁腈、仲丁醇、正丙醇、正戊醇或异戊醇中的一种或多种;优选地,步骤2中所述第二溶剂选自甲醇。
[0051]
在部分实施方案中,步骤2中所述第二溶剂的加入量为使步骤2中的氢溴酸浓度稀释1-40倍;优选稀释2倍。
[0052]
在部分实施方案中,步骤2中所述氢溴酸初始浓度是1mol/l的氢溴酸水溶液或者质量浓度为48%氢溴酸水溶液。
[0053]
在部分实施方案中,步骤3中所述温度为25-50℃,优选30℃;所述滴加速率为0.5-10ml/min,优选2ml/min;
[0054]
在部分实施方案中,步骤4中所述降温速率为0.1~2℃/min,优选降温速率为0.5℃/min;所述干燥的条件为20~55℃、真空度0.08mpa~0.1mpa的条件下进行2-10h。
[0055]
药物组合物和治疗方法
[0056]
本发明第三方面提供一种药物组合物,其包含本发明式(i)化合物的氢溴酸盐晶型i,以及一种或多种药学上可接受的载体。
[0057]
所述“药学上可接受的载体”是指与治疗剂一同给药的稀释剂、辅剂、赋形剂或媒介物,并且其在合理的医学判断的范围内适于接触人类和/或其它动物的组织而没有过度的毒性、刺激、过敏反应或与合理的益处/风险比相应的其它问题或并发症。
[0058]
本发明第四方面提供式(i)化合物的氢溴酸盐晶型i和/或本发明的药物组合物在制备用于预防和/或治疗糜烂性食管炎、胃溃疡、十二指肠溃疡、幽门螺杆菌根除及胃酸过多引起的相关疾病的药物中的用途。
[0059]
本发明第五方面提供一种预防和/或治疗糜烂性食管炎、胃溃疡、十二指肠溃疡、幽门螺杆菌根除及胃酸过多引起的相关疾病的方法,所述方法包括向需要其的个体给药有效量的本发明式(i)化合物的氢溴酸盐晶型i和/或本发明的药物组合物。
[0060]
本发明提供的式(i)化合物的氢溴酸盐晶型i,具有以下有益效果:
[0061]
(1)结晶度好,纯度高,具有良好的热稳定性,并且工艺安全性高,操作简单,产品形貌呈“棒状”大粒径晶体,后处理简单,晶体纯度高,无溶剂残留问题,适于工业生产;
[0062]
(2)溶解性好,较现有技术中的富马酸盐的晶型相比,溶解度高5-10倍,溶解速度快,有利于新剂型的开发;
[0063]
(3)引湿性更低,有利于后续制剂的开发以及存放、运输。
附图说明
[0064]
图1:式(i)化合物的氢溴酸盐晶型i的xrpd图谱;
[0065]
图2:式(i)化合物的氢溴酸盐晶型i的dsc图谱;
[0066]
图3:式(i)化合物的氢溴酸盐晶型i的tga图谱;
[0067]
图4:式(i)化合物的氢溴酸盐晶型i的plm图谱(50倍);
[0068]
图5:式(i)化合物不同盐的dvs图谱;
[0069]
图6:式(i)化合物醋酸盐晶型的xrpd图谱;
[0070]
图7:式(i)化合物氢溴酸盐晶型i光照条件下的xrpd图谱;
[0071]
图8:式(i)化合物氢溴酸盐晶型i加速条件下的(40℃/rh75%)xrpd图谱;
[0072]
图9:式(i)化合物氢溴酸盐晶型i高湿条件下的(40℃/rh92.5%)xrpd图谱;
[0073]
图10:式(i)化合物氢溴酸盐晶型i高温条件下的(60℃)xrpd图谱;
[0074]
图11:式(i)的化合物马来酸盐晶型a的xrpd图谱;
[0075]
图12:式(i)的化合物马来酸盐晶型a的plm图谱;
[0076]
图13:式(i)的化合物马来酸盐晶型b的xrpd图谱;
[0077]
图14:式(i)的化合物马来酸盐晶型b的plm图谱;
[0078]
图15:式(i)的化合物丁二酸盐晶型a的xrpd图谱;
[0079]
图16:式(i)的化合物丁二酸盐晶型a的plm图谱;
[0080]
图17:式(i)的化合物磷酸盐晶型a的xrpd图谱;
[0081]
图18:式(i)的化合物磷酸盐晶型a的plm图谱。
具体实施方式
[0082]
以下通过实施例对本发明做进一步阐述,本发明的实施例仅用于说明本发明的技术方案,并非用于限定本发明的范围,本领域技术人员可进行一些非本质的改进和调整,仍属于本发明的保护范围。
[0083]
实验所用的测试仪器信息和方法:
[0084]
x-射线粉末衍射(xrpd):
[0085]
采用x`pert3 powder diffractometer,该仪器采用cu靶照射,在室温下使用absolute scan进行检测。检测范围在3.5
°
至40
°
,步长为0.013,停留时间为50s,扫描1次。
[0086]
差示扫描热量法(dsc)测试仪器为:ta dsc 2500;
[0087]
热重分析(tga)测试仪器为:mettler toledo;
[0088]
dsc和tga仪器的加热速度均为10k/min。
[0089]
动态水份吸附仪(dvs)实验条件如下:
[0090]
采用dvs intrinsic(sms),在25℃,dmdt模式下进行检测。
[0091]
核磁nmr的测定使用bruker核磁共振仪,仪器型号:brbker avanceⅲhd400mhz;测试条件:溶剂dmso-d6,温度23.5℃。
[0092]
偏光显微镜(plm)检测的仪器为:尼康elipse光学显微镜,放大50倍观察晶体形貌。
[0093]
伏诺拉生氢溴酸盐或其晶型中溴含量的检测方法实验条件如下:
[0094]
照离子色谱法(中国药典2020年版四部通则0513)测定。
[0095]
供试品溶液:取本品适量,精密称定,加水溶解并稀释成每1ml中约含6-300μg的溶液;对照品溶液:精密量取溴单元素标准溶液适量,用水定量稀释成每1ml中含1-50μg的溶液。
[0096]
色谱条件:阴离子交换色谱柱;检测器为电导检测器;检测方式为抑制电导检测;淋洗液:oh-体系-5-40mmol/l;柱温:20-40℃;进样量:10-200μl。
[0097]
系统适用性要求:供试品溶液色谱图中,溴与相邻峰之间的分离度应符合要求。
[0098]
精密量取供试品溶液和对照品溶液,分别注入离子色谱仪,记录色谱图。按外标法
以峰面积计算。
[0099]
制备例1:伏诺拉生游离碱的制备
[0100]
按照专利cn105524046 a实施例5的方法制备得到伏诺拉生游离碱,对制得的伏诺拉生游离碱进行hplc检测,其纯度为99.55%,将该制备例制得的化合物用于以下实施例。
[0101]
实施例1氢溴酸盐晶型i的制备
[0102]
称量1.7g的式(i)化合物(游离碱),加入20ml甲醇中,升温至50℃溶清;称量995mg质量浓度为48%的溴化氢水溶液,加入20ml甲醇稀释,然后将稀释后的氢溴酸溶液以2ml/min的速率滴加至游离碱的溶清液中。滴加完毕未析晶,自然降温至室温后析晶。收集固体,20ml甲醇洗涤,40℃烘箱、真空度0.08mpa、干燥7h,得到白色固体即为本发明的式(i)的化合物氢溴酸盐晶型i。检测分析其xrpd图谱如图1示,dsc和tga图谱分别如图2和图3所示。
[0103]
所得固体产品的核磁数据如下:1h nmr(400mhz,dmso-d6)δ8.93-8.90(m,3h),8.58(d,j=2.07hz,1h),7.94-7.91(m,1h),7.88(d,j=1.76hz,1h),7.68-7.64(m,1h),7.58-7.52(m,1h),7.28-7.23(m,2h),7.13-7.09(m,1h),6.60(d,j=1.66hz,1h),4.05(s,2h),2.56(s,3h)。
[0104]
所得白色固体产品中br含量为18.1%,故式(i)的化合物氢溴酸盐晶型i中式(i)化合物与氢溴酸的摩尔比为1:1。
[0105]
实施例2氢溴酸盐晶型i的制备
[0106]
称量34g的式(i)的化合物(游离碱),加入400ml丙酮中,升温至50℃溶清,溶清后自然降温至室温;称量19.91g质量浓度为48%的溴化氢水溶液,加入200ml甲醇稀释,然后将稀释后的氢溴酸溶液以10ml/min的速率滴加至游离碱的溶清液中。滴加过程中析出固体,滴加完毕养晶30min后,以0.5℃/min降温至5℃,继续养晶0.5-1h后收集固体。滤饼用400ml丙酮洗涤,30℃烘箱、真空度0.1mpa、干燥10h,得到白色固体。检测分析其xrpd图谱、dsc、tga图谱均与实施例1一致,说明其为式(i)的化合物氢溴酸盐晶型i;其plm显微图片如图4所示。
[0107]
实施例3氢溴酸盐晶型i的制备
[0108]
称量30g的式(i)的化合物(游离碱),加入250ml乙酸乙酯中,升温至40℃溶清;称量17.56g质量浓度为48%的溴化氢水溶液,加入15ml甲醇稀释,然后将稀释后的氢溴酸溶液以0.5ml/min的速率滴加至游离碱的溶清液中。滴加完毕析晶,自然降温至室温后收集固体,200ml乙酸乙酯洗涤,50℃烘箱、真空度0.1mpa、干燥2h,得到白色固体。检测分析其xrpd图谱、dsc、tga图谱均与实施例1一致,说明其为式(i)的化合物氢溴酸盐晶型i。
[0109]
对比例1醋酸盐晶型的制备
[0110]
称量5.09g的式(i)的化合物(游离碱),加入20ml乙酸乙酯中,室温下得到悬浮液;称量0.93g醋酸溶液,加入20ml乙酸乙酯稀释后,将其缓慢滴加至游离碱的悬浮液中,滴加过程中变为澄清的黄色澄清液,降温至15℃,恒定至15℃析晶,析晶得到白色固体后,继续养晶24h后,收集固体,20ml乙酸乙酯洗涤,50℃烘箱、真空度0.1mpa、干燥2h,得到白色固体。检测分析其xrpd图谱如图6所示,经分析,其为醋酸盐已知晶型a和晶型c的混晶。
[0111]
对比例2采用乙醇制备伏诺拉生氢溴酸盐
[0112]
称量1.7g的式(i)化合物(游离碱),量取20ml乙醇;称量1.99g质量浓度为48%的溴化氢水溶液,采用所有物料室温直接混合的方式,成盐析晶,搅拌速度250r/min,成盐反
应时间5h后得到的固体,收集固体,20ml乙醇洗涤,50℃烘箱、真空度0.1mpa、干燥3h,得到固体。其为二氢溴酸盐,经xrpd测定,其结晶度不高,hplc测定纯度为99.5%。
[0113]
对比例3采用乙酸乙酯制备伏诺拉生氢溴酸盐
[0114]
称量1.7g的式(i)的化合物(游离碱),量取20ml乙酸乙酯;称量1.99g质量浓度为48%的溴化氢水溶液,采用所有物料室温直接混合的方式,成盐析晶,搅拌速度400r/min,成盐反应时间3h后收集固体,20ml乙酸乙酯洗涤,50℃烘箱、真空度0.1mpa、干燥2h,得到白色固体。经xrpd分析得到的固体为式i化合物氢溴酸盐的无定形,纯度99.5%。
[0115]
对比例4马来酸盐晶型a的制备
[0116]
称量1.7g式(i)化合物(游离碱形式),加入20ml乙腈中,搅拌溶清。称量马来酸70.50mg,加入乙腈20ml、纯化水2ml溶解得到马来酸溶液。将马来酸溶液在60℃下,以0.5ml/min速度缓慢滴加至式(i)的化合物的溶清液中,搅拌2h,搅拌速度250r/min,利用0.25μm的滤膜热过滤得到澄清液,室温下仍为澄清,开始降温,降温速率0.5℃/min,从室温降温至3℃,收集固体,在25℃真空烘箱干燥24h得到固体,为马来酸盐晶型a。经检测,其xrpd图谱如图11所示,plm图谱如图12所示,dvs引湿性结果如图5所示。
[0117]
对比例5马来酸盐晶型b的制备
[0118]
称量0.17g式(i)化合物(游离碱形式),加入2ml丙酮,磁力搅拌溶清。称量马来酸70.11mg,加入至式(i)化合物游离碱溶清液中,磁力搅拌反应2h得白色固体,离心收集固体,在25℃真空烘箱干燥24h,得到固体,为马来酸盐晶型b。经检测,其xrpd图谱如图13所示,plm图谱如图14所示。
[0119]
对比例6丁二酸盐晶型a的制备
[0120]
称量0.17g式(i)化合物(游离碱形式),加入2ml甲醇,磁力搅拌溶清。称量丁二酸72.0 5mg固体,用1ml甲醇溶解后,60℃下缓慢滴加至式(i)化合物游离碱溶清液中,磁力搅拌反应2h,5℃-40℃循环升降温10h,得白色固体,离心收集固体,在25℃真空烘箱干燥24h,得到固体,为丁二酸盐晶型a。经检测,其xrpd图谱如图15所示,plm图谱如图16所示。
[0121]
对比例7磷酸盐晶型a的制备
[0122]
称量0.17g式(i)化合物(游离碱形式),加入2ml乙酸乙酯,40℃磁力搅拌溶清。称量69.00mg质量浓度为85%的磷酸水溶液,加入1ml乙酸乙酯稀释后,成滴状缓慢滴加至式(i)化合物游离碱溶清液中,磁力搅拌反应2h,5℃-40℃循环升降温16h,得白色固体,离心收集固体,在25℃真空烘箱干燥24h,得到固体,为磷酸盐晶型a。经检测,其xrpd图谱如图17所示,plm图谱如图18所示。
[0123]
对比例8对甲苯磺酸盐晶型a的制备
[0124]
称量0.17g式(i)化合物(游离碱形式),加入2ml异丙醇,40℃磁力搅拌溶清。称量10.31mg对甲苯磺酸固体,加入1ml纯化水溶解后,缓慢成滴加入至式(i)化合物游离碱溶清液中,磁力搅拌反应2h,5℃-40℃循环升降温20h,离心收集固体,在25℃真空烘箱干燥24h,得白色固体,为对甲苯磺酸盐晶型a,经检测,其dvs引湿性如图5所示。
[0125]
对比例9l-苹果酸盐晶型a的制备
[0126]
称量0.17g式(i)化合物(游离碱形式),加入2ml 1,4-二氧六环,磁力搅拌溶清。称量l-苹果酸盐80.45mg,加至式(i)化合物游离碱溶清液中,磁力搅拌反应10h得白色固体,离心收集固体,在25℃真空烘箱干燥24h,得到固体,为l-苹果酸盐晶型a。经检测,其dvs引
湿性如图5所示。
[0127]
试验例1溶解度考察
[0128]
待测样品:
[0129]
富马酸伏诺拉生晶型a:按照专利cn200680040789.7方法制备得到,纯度为99.9%。
[0130]
氢溴酸伏诺拉生晶型i:实施例2制备得到,纯度99.99%。
[0131]
室温纯水中溶解度试验方法:称量50.00g纯化水,控制温度为20-21℃,采用少量多次的加入待测样品直至不再溶解为止,累计加入的待测样品固体总量计算其溶解度,测定结果如下表所示。
[0132]
高温纯水中溶解度的试验方法:将50ml纯化水升温并控制在70℃,采用少量多次的加入待测样品直至不再溶解为止,累计加入的待测样品固体总量计算其溶解度,测定结果如下表所示。
[0133]
表1溶解度考察结果
[0134]
待测样品20-21℃水溶解度70℃水溶解度富马酸伏诺拉生晶型a2.95mg/ml<5mg/ml氢溴酸伏诺拉生晶型i10.8mg/ml10-20mg/ml
[0135]
溶解性数据表明,氢溴酸盐晶型i较富马酸盐晶型a的溶解度大幅提高,且实验过程中,观察氢溴酸盐溶解速度非常快,有利于制剂剂型的选择及工业化放大生产,同时有利于药物在体内药效的发挥。
[0136]
试验例2不同盐型的dvs引湿性考察
[0137]
待测样品:对比例8制备的对甲苯磺酸盐晶型a、对比例9制备的l-苹果酸盐晶型a、对比例4制备的马来酸盐晶型a、对比例6制备的丁二酸盐晶型a、实施例2制备得到的氢溴酸盐晶型i。
[0138]
将待测样品置于25℃真空烘箱中干燥48h后,利用dvs动态水分吸附仪(dvs intrinsic)测试其25℃条件下的引湿性,测试条件,rh%湿度步长10%,测试湿度区间rh 0-90%,dm/dt模式平衡。
[0139]
不同盐型的dvs引湿性考察数据如图5所示,从上述dvs测定结果可以看出,本专利的氢溴酸盐晶型i相比于l-苹果酸盐晶型a、马来酸盐晶型a、丁二酸盐晶型a、对甲苯磺酸盐晶型a,其引湿性最低,有利于原料api的保存、运输,以及制剂工艺的操作。
[0140]
试验例3稳定性考察
[0141]
将实施例2制备得到的氢溴酸盐晶型i分别放置于光照(5000lx)、高温60℃、高湿40℃/rh=92.5%、加速40℃/rh=75%四种条件下,分别于11天、32天考察其化学稳定性和晶型稳定性的变化情况。其中,采用xrpd和dsc测定晶型变化情况,hplc测定化学稳定性变化情况,考察结果如下表所示。
[0142]
表2稳定性考察结果
[0143][0144]
稳定性考察结果表明,本发明的氢溴酸盐晶型i在高温、高湿及加速条件下,化学纯度及晶型均未发生明显变化,说明化学稳定性及晶型稳定性均较好。
[0145]
试验例4氢溴酸伏诺拉生和富马酸伏诺拉生对注射剂质量的影响
[0146]
表3处方组成
[0147]
处方1#2#氢溴酸伏诺拉生1.0g-富马酸伏诺拉生-1.0g氯化钠0.9g0.9g枸橼酸钠-适量注射用水100ml100ml
[0148]
1#注射剂:量取适量的注射用水,加热到70℃,按照上表处方用量,依次加入处方量的氯化钠搅拌溶解,再加入处方量的氢溴酸伏诺拉生晶型i(实施例2)搅拌溶解,凉至室温,检测ph4.2,补水至全量,灌封,将样品于121℃/12min条件下灭菌,得到1#注射剂。
[0149]
2#注射剂:量取适量的注射用水,加热到70℃,按照上表处方用量,依次加入处方量的氯化钠搅拌溶解,加入处方量的富马酸伏诺拉生晶型a搅拌,因为其不能完全溶解,需调解ph≥6.5纯水条件下富马酸伏诺拉生溶解度才能达到10-15mg/ml;故用枸橼酸钠溶液调节ph至6.51,搅拌溶解,补水至全量,灌封,将样品于121℃/12min条件下灭菌,得到2#注射剂。
[0150]
将上述注射剂分别于灭菌前后取样考察其杂质情况,并且将灭菌后的样品放置于60℃条件下10天,取样测定其杂质含量变化,测定结果如下表所示。
[0151]
表4有关物质考察结果
[0152][0153]“/”表示未检出。
[0154]
从以上数据可以看出,采用富马酸伏诺拉生作为原料药制备得到的注射液杂质个数及总杂质含量均明显大于氢溴酸伏诺拉生注射液。富马酸伏诺拉生溶解性很差,需在偏
碱性条件下溶解度才能增加至满足常规注射剂开发的要求,而杂质yz2-2受ph影响,ph越偏碱性,增长越明显,这就注定了富马酸盐伏诺拉生满足了溶解性,质量稳定性就会存在问题,不利于液体制剂开发;而氢溴酸伏诺拉生由于显著改善了溶解性的问题,因此符合液体制剂开发的需要。
[0155]
本技术公开的式(i)化合物的固体形式及其制备方法,本领域技术人员可通过借鉴本文内容,适当改变原料、工艺参数等环节实现。本技术的方法与产品已通过较佳实施例进行了描述,相关技术人员明显能在不脱离本技术内容、精神和范围内对本文所述的方法和产品进行改动或适当变更与组合,来实现本技术技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本技术精神、范围和内容中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。