1.本发明属于药物化学技术领域,具体涉及一种非奈利酮及其中间体的制备方法。
2.背景领域
3.非奈利酮(英文名:finerenone),是由拜耳公司开发的一种充当盐皮质激素受体的非甾体拮抗剂,并且可用作预防和/或治疗心血管和肾脏疾病例如心力衰竭和糖尿病肾病的药剂,化学名为(4s)-4-(4-氰基-2-甲氧基苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺。非奈利酮结构式如下:
[0004][0005]
目前有多个文献报道了非奈利酮的制备方法,其中wo2016016287公开的制备方法如路线1所示。
[0006][0007]
wo2017032673公开的制备方法如路线2所示。
[0008][0009]
路线1和路线2所述工艺均在得到非奈利酮消旋体后才进行拆分,总收率上不经济,而且采用的均为手性hplc拆分方法,此方法生产成本非常高,不适合工业化大生产。后期有wo2021074072和wo2021074078分别报道了采用拆分剂拆分中间体的工艺,分别如路线3和路线4所示:
[0010]
[0011][0012]
路线3拆分得到中间体非对映体盐de值小于85%,经过进一步纯化后非对映体盐de值提高到98%,加碱游离后得到的中间体a的ee值小于99%,拆分总收率小于40%。路线4拆分得到中间体非对映体盐de值小于80%,经过进一步纯化后非对映体盐de值提高到99%,加碱游离后得到的中间体b的ee值99%,拆分总收率40%左右。两条路线得到中间体非对映体盐de值均不高,都需要进一步纯化提高ee值。
[0013]
因此,本领域迫切需要开发一种收率高、操作简便、质量控制更好的、适合工业化生产的制备非奈利酮的工艺。
技术实现要素:
[0014]
本发明所要解决的技术问题就是针对现有技术中非奈利酮的制备存在的外消旋体拆分成本高、产品ee值低、不适合工业化生产等问题,而提出了一种非奈利酮的制备方法。
[0015]
在本发明的第一方面,提供了一种制备式ⅰ化合物的方法,包括步骤:
[0016]
(1)将外消旋体式ⅱ化合物与式ⅲa化合物或式ⅲb化合物所示的拆分剂混合,进行成盐反应,经分离分别得到式ⅳa化合物或式ⅳc化合物所示的盐;
[0017]
(2)将步骤1得到的式ⅳa化合物或式ⅳc化合物所示的盐用碱处理,得到式ⅰ化合物;
[0018]
反应式如下:
conhr、-conrr’,其中所述各个r、r’各自独立地为甲基、乙基或苯基,或n、r、r’与其相连的碳原子共同构成5-7元的氮杂环。
[0027]
在另一优选例中,ar为未取代或取代的c10-c14的多环芳基,例如萘基、蒽基。
[0028]
在另一优选例中,ar为未取代或取代的c5-c10杂芳基,例如哌啶基、哌嗪基、喹啉基。
[0029]
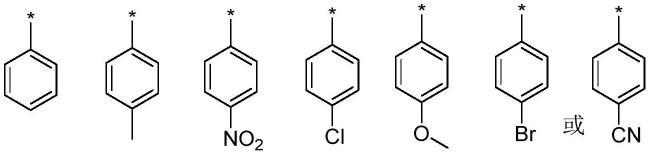
在另一优选例中,ar选自下组:
[0030][0031]
其中*代表连接点。
[0032]
在另一优选例中,ar为单取代苯基。
[0033]
在另一优选例中,ar为对位取代苯基。
[0034]
在另一优选例中,ar为苯甲基、苯基、硝基苯基、氯苯基、溴苯基、苯甲氧基、氰基苯基。
[0035]
在另一优选例中,ar为苯甲基、苯基、苯甲氧基。
[0036]
在另一优选例中,ar为苯基或苯甲基。
[0037]
在另一优选例中,ar为苯甲基。
[0038]
在另一优选例中,在步骤1中,式ⅱ化合物与式ⅲa化合物的摩尔比为1:0.4-1.2,优选1:0.6-1.2,更优选1:1。
[0039]
在另一优选例中,在步骤1中,式ⅱ化合物与式iiib化合物的摩尔比为1:0.4-1.2,优选1:0.6-1.2,更优选1:1。
[0040]
在另一优选例中,步骤1中,所述拆分剂为式iiib化合物。
[0041]
在另一优选例中,步骤1中,所述成盐反应在有机溶剂或水与有机溶剂的混合溶剂中进行,其中所述的有机溶剂选自丙酮、2-丁酮、甲基异丁基酮、乙酸乙酯、二氯甲烷、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚或二氧六环或其组合。
[0042]
在另一优选例中,步骤1中,所述成盐反应还具有以下的一个或多个特征:
[0043]
(a)所述式ii化合物在有机溶剂中的浓度为0.1-0.3mmol/ml,优选0.14-0.23mmol/ml;
[0044]
(b)所述成盐反应在60-70℃下反应;
[0045]
(c)所述成盐反应的反应时间为2-4h,优选3h。
[0046]
在另一优选例中,步骤1所述的分离为沉淀分离。
[0047]
在另一优选例中,在步骤1中,所述分离包括:将反应体系冷却至室温,搅拌12-18h析出,过滤分离。
[0048]
在另一优选例中,过滤分离得到的固体为式ⅳa化合物或式ⅳc化合物,滤液中的式ⅳb化合物或式ⅳd化合物可通过碱处理后,再用wo2017032678公开的方法进行非对映异构体的回收。
[0049]
在另一优选例中,在步骤2中,所述碱为氢氧化钾、磷酸钾或磷酸钠。
[0050]
在另一优选例中,步骤2的碱处理步骤在水、有机溶剂或水与有机溶剂的混合溶剂
中进行,其中所述的有机溶剂选自丙酮、2-丁酮、甲基异丁基酮、乙酸乙酯、二氯甲烷、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚或二氧六环或其组合。
[0051]
在另一优选例中,在步骤2中,所述碱处理在碱的水溶液中进行。
[0052]
在另一优选例中,在步骤2中,所述碱处理步骤的ph为7-7.5。
[0053]
在另一优选例中,步骤2中,所述碱处理还具有以下的一个或多个特征:
[0054]
(a)所述式ⅳa化合物或式ⅳc化合物所示的拆分盐在溶剂中的浓度各自独立地为0.05-0.15g/ml;
[0055]
(b)所述碱处理在15-25℃下反应;
[0056]
(c)所述碱处理的反应时间为2-4h,优选3h。
[0057]
在另一优选例中,式ⅱ化合物由以下步骤制备得到:
[0058]
i)化合物1与3-氧代丁酸2-苄酯反应得到化合物2与化合物2’的混合物;
[0059]
ii)将步骤i得到的混合物与4-氨基-5-甲基吡啶酮反应得到化合物3;
[0060]
iii)化合物3在浓硫酸催化下与原甲酸三乙酯反应得到式ⅱ化合物;
[0061]
反应式如下:
[0062][0063]
在本发明的第二方面,提供了一种制备非奈利酮的方法,所述方法包括如下步骤:
[0064]
(s1)提供式ii化合物;
[0065]
(s2)以式ii化合物为原料,经拆分,制备得到式i化合物,其中所述制备式i化合物的方法如本发明第一方面所述;
[0066]
(s3)式ⅰ化合物经还原得到式
ⅰ‑
1化合物;
[0067]
(s4)式i-1化合物经氨化得到非奈利酮;
[0068]
反应式如下:
[0069]
[0070]
在另一优选例中,在步骤(s1)中,所述式ii化合物由以下步骤制备得到:
[0071]
i)化合物1与3-氧代丁酸2-苄酯反应得到化合物2与化合物2’的混合物;
[0072]
ii)将步骤i得到的混合物与4-氨基-5-甲基吡啶酮反应得到化合物3;
[0073]
iii)化合物3在浓硫酸催化下与原甲酸三乙酯反应得到式ⅱ化合物;
[0074]
反应式如下:
[0075][0076]
在本发明的第三方面,提供了一种非对映体盐或其药学上可接受的盐,如下式所示:
[0077][0078]
其中,ar如本发明第一方面所定义。
[0079]
在另一优选例中,所述非对映体盐为iva或ivc。
[0080]
在另一优选例中,所述非对映体盐为ivc。
[0081]
在另一优选例中,所述非对映体盐选自iva-1、iva-2、ivc-1~ivc-6中的任一非对映体盐。
[0082]
在本发明的第四方面,提供了一种非奈利酮中间体,所述中间体为式ii化合物或其异构体或其药学上可接受的盐
[0083][0084]
在另一优选例中,所述中间体为外消旋体式ii化合物。
[0085]
在另一优选例中,所述中间体为式ii化合物的(s)-异构体,即式i化合物
[0086][0087]
在本发明的第五方面,提供了一种本发明第三方面所述的非对映体盐或其药学上可接受的盐,和本发明第四方面所述的式ii化合物或其对映异构体或其药学上可接受的盐的用途,用于作为非奈利酮制备过程中的中间体,合成非奈利酮。
具体实施方式
[0088]
本发明人经过广泛而深入的研究,首次意外地发现了一种制备超高ee值非奈利酮的方法,具体地,发明人发现使用特定的被拆分中间体,将被拆分中间体的氰乙基替换成苄基后仅需简单分离,无需进一步纯化即可得到de值超过98.5%的非对映体盐,接着加碱游离后得到的中间体的ee值高达99.8%,最终得到ee值超过99.9%的非奈利酮。本方法操作简便、收率高、产品纯度高,非常适合工业化生产制备非奈利酮。在此基础上,发明人完成了本发明。
[0089]
非奈利酮中间体
[0090]
本发明提供了一种非奈利酮中间体,所述中间体为外消旋的式ii化合物或其药学上可接受的盐;
[0091][0092]
其中,表示为外消旋结构。
[0093]
本发明还提供了一种非奈利酮中间体,所述的中间体为式i化合物,即式ii化合物拆分得到的(s)-异构体。
[0094][0095]
本发明中拆分剂的羧基与式ⅱ化合物中二氢吡啶环的二级胺基形成非对映体拆分盐。二氢吡啶环是一个刚性的平面结构,因此连接手性中心的酯基的位阻对拆分效果应该不会有显著影响。
[0096]
但令人惊奇的是,本发明将被拆分化合物的氰乙基替换成苄基(式ii化合物)后,却发现在拆分反应过程中非对映体盐仅经过简单分离、不需要进一步的纯化,其de值即可达到98.5%以上。
[0097]
制备非奈利酮中间体式ⅰ化合物的方法
[0098]
本发明提供了一种制备非奈利酮中间体式ⅰ化合物的方法,包括步骤:
[0099]
(1)将外消旋体式ⅱ化合物与式ⅲa化合物或式ⅲb化合物所示的拆分剂的反应,经分离分别得到式ⅳa化合物或式ⅳc化合物所示的盐;
[0100]
(2)将步骤1得到的盐进行碱析,得到式ⅰ化合物。
[0101]
反应式如下:
[0102][0103]
其中,ar代表未取代或取代的芳族或杂芳族基团,优选的ar代表:
[0104][0105]
其中r1、r2、r3、r4、r5各自代表氢原子或烷基,例如甲基、乙基、丙基;或
[0106]
卤素原子,例如氟、氯、溴或碘;或
[0107]
醚基,例如o-甲基、o-乙基、o-苯基;或
[0108]
硝基、氰基、cf3基或
[0109]
酰胺基,所述酰胺基为例如nhcor,其中r可代表甲基、乙基或苯基,或-nrcor基团,其中r具有上述含义,或conhr基团,其中r具有上述含义,或conrr’基团,其中r’具有与如上所定义的r相同的含义,或代表环酰胺如-co-哌啶基。取代方式可能有很大的差异。
[0110]
因此,理论上可能有最高达5个不同的取代基,但通常优选单取代的ar基团。
[0111]
然而,ar也可为取代的杂芳族基团,例如优选哌啶或哌嗪。
[0112]
也可为多环芳烃,例如取代的萘、蒽或喹啉。
[0113]
特别优选的ar为:
[0114][0115]
其中*代表连接点。
[0116]
更特别优选的ar为优选为苯基或甲基苯。
[0117]
在另一优选例中,步骤1中,式ⅱ化合物与式ⅲa化合物或式ⅲb化合物的摩尔比为1:0.4-1.2,优选1:0.6-1.2。
[0118]
在另一优选例中,步骤1的成盐反应在有机溶剂或水与有机溶剂的混合溶剂中进行,所述的有机溶剂选自丙酮、2-丁酮、甲基异丁基酮、乙酸乙酯、二氯甲烷、四氢呋喃、二甲基四氢呋喃、乙二醇二甲醚或二氧六环或其组合。
[0119]
在另一优选例中,步骤1所述的分离为沉淀分离。
[0120]
过滤得到的固体为式ⅳa化合物或式ⅳc化合物,滤液中的式ⅳb化合物或式ⅳd化合物可通过碱处理后,再用wo2017032678公开的方法进行非对映异构体的回收。
[0121]
在另一优选例中,步骤2所述的碱为氢氧化钾、磷酸钾或磷酸钠。
[0122]
在另一优选例中,步骤2的碱处理步骤在水、有机溶剂或水与有机溶剂的混合溶剂中进行,所述的有机溶剂选自丙酮、2-丁酮、甲基异丁基酮、乙酸乙酯、二氯甲烷、四氢呋喃、二甲基四氢呋喃、乙二醇二甲醚或二氧六环或其组合。
[0123]
一种制备非奈利酮的方法
[0124]
本发明提供了一种制备非奈利酮的方法,本发明使用式ii化合物作为被拆分中间体,经过简单的拆分即可得到高ee值的式i化合物,用于制备非奈利酮。
[0125]
本发明所述的制备非奈利酮的方法包括以下步骤:
[0126]
(s1)提供式ii化合物;
[0127]
(s2)以式ii化合物为原料,经拆分,制备得到式i化合物,其中所述制备式i化合物的方法如本发明所述;
[0128]
(s3)式ⅰ化合物经钯炭还原得到式
ⅰ‑
1化合物;
[0129]
(s4)式
ⅰ‑
1化合物经氨化得到非奈利酮。
[0130]
反应式如下:
[0131][0132]
本发明的制备方法优势在于:
[0133]
(a)反应步骤简洁,反应条件温和,无需特殊的试剂,操作简便,适合工业化生产;
[0134]
(b)通过酒石酸酯来进行对映体混合物的分离,可以避免使用高成本、低效率的手
性相色谱方法分离,且通过酒石酸酯分离对映体混合物的收率在65%以上,中间体非对映体盐的de值98.5%以上,中间体ee值可达99.5%以上,最终产品非奈利酮的ee值可达99.8%以上。
[0135]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0136]
除非特别说明,实施例中所用的原料或试剂均可通过市售获得或者按照常规方法制备。
[0137]
实施例中所用的原料4-甲酰基-3-甲氧基苄腈可通过wo2017032673公开的方法制备得到。
[0138]
实施例1化合物3的制备
[0139][0140]
在分水器上,将4-甲酰基-3-甲氧基苄腈(30.6g,189.87mmol)、3-氧代丁酸2-苄酯(45.6g,237.34mmon)、哌啶(1.9ml)和冰醋酸(2.2ml)于二氯甲烷(300ml)中,在回流条件下反应18h。使混合物冷却至室温,并将有机相每次用水洗涤两次。然后浓缩二氯甲烷,得到粘稠状液体,溶于2-丁醇(380ml)中,并加入4-氨基-5-甲基吡啶酮(20.8g,167.13mmol),升温至回流搅拌20h。冷却至0℃并在该温度下搅拌4h,过滤,用2-丁醇(100ml)漂洗。在40℃条件下真空干燥16h,得到淡黄色固体63.5g,收率86.0%(基于4-氨基-5-甲基吡啶酮计算),hplc纯度99.5%。
[0141]
实施例2式ⅱ化合物的制备
[0142][0143]
将4-(4-氰基-2-甲氧基苯基)-2,8-二甲基-5-氧代-1,4,5,6-四氢-1,6-萘啶-3-甲酸2-苄酯(40.0g,90.6mmol)和原甲酸三乙酯(268.6g,1812.1mmol)溶于n,n-二甲基乙酰胺(1000ml)中,并加入浓硫酸(1.2ml)。将混合物在115℃下加热2h,然后冷却室温,减压浓缩n,n-二甲基乙酰胺,加入二氯甲烷(1000ml)和水(1000ml),搅拌后静止分层,浓缩有机相
然后加入甲醇(500ml)打浆得到淡黄色固体38.3g,收率90.1%,hplc纯度99.8%。1h nmr(400mhz,[d6]dmso):δ=1.05(t,3h),2.12(s,3h),2.18(s,3h),3.82(s,3h),3.96
–
4.06(m,2h),5.37(s,1h),6.49
–
6.86(2h),7.15(d,1h),7.28(d,1h),7.30(m,5h),7.37(d,1h),7.55(s,1h),7.68(s,1h)。ms:m/z=470[m 1]
。
[0144]
实施例3式ⅳa-1的制备
[0145][0146]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲa-1化合物(4.1g,10.6mmol)(式ⅲa化合物中ar为苯甲基),甲基异丁基酮(75ml),然后加热至内温70℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用甲基异丁基酮(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.7g(理论值的104.0%),de值99.0%。
[0147]
实施例4式ⅳa-2的制备
[0148][0149]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲa-2化合物(3.8g,10.6mmol)(式ⅲa化合物中ar为苯基),乙二醇二甲醚(75ml),然后加热至内温70℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用乙二醇二甲醚(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.3g(理论值的98.0%),de值99.2%。
[0150]
实施例5式ⅳc-1的制备
[0151][0152]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-1化合物(4.1g,10.6mmol)(式ⅲb化合物中ar为苯甲基),四氢呋喃(75ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用四氢呋喃(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.7g(理论值的104.0%),de值99.1%。
[0153]
实施例6式ⅳc-2的制备
[0154][0155]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-2化合物(4.8g,10.6mmol)(式ⅲb化合物中ar为硝基苯),四氢呋喃(50ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用四氢呋喃(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.9g(理论值的100.0%),de值98.8%。
[0156]
实施例7式ⅳc-3的制备
[0157][0158]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-3化合
物(4.5g,10.6mmol)(式ⅲb化合物中ar为氯苯基),丙酮(45ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用丙酮(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.8g(理论值的100.0%),de值99.0%。
[0159]
实施例8式ⅳc-4的制备
[0160][0161]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-4化合物(2.7g,6.4mmol)(式ⅲb化合物中ar为苯甲氧基),乙酸乙酯(75ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用乙酸乙酯(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.7g(理论值的100.0%),de值99.2%。
[0162]
实施例9式ⅳc-5的制备
[0163][0164]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-5化合物(6.6g,12.7mmol)(式ⅲb化合物中ar为溴苯基),二氯甲烷(75ml),然后加热至内温35℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用二氯甲烷(2.5ml)漂洗固体,45℃真空干燥,得到白色固体5.0g(理论值的95.0%),de值99.2%。
[0165]
实施例10式ⅳc-6的制备
[0166][0167]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-6化合物(4.3g,10.6mmol)(式ⅲb化合物中ar为氰基苯),四氢呋喃(75ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用四氢呋喃(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.6g(理论值的98.9%),de值99.4%。
[0168]
实施例11式ⅳc-1的制备
[0169][0170]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-1化合物(4.1g,10.6mmol)(式ⅲb化合物中ar为苯甲基),2-甲基四氢呋喃(75ml),然后加热至内温60℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用2-甲基四氢呋喃(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.6g(理论值的101.1%),de值99.5%。
[0171]
实施例12式ⅳc-1的制备
[0172]
[0173]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-1化合物(4.1g,10.6mmol)(式ⅲb化合物中ar为苯甲基),乙二醇二甲醚(100ml),然后加热至内温70℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用乙二醇二甲醚(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.5g(理论值的98.9%),de值99.4%。
[0174]
实施例13式ⅳc-1的制备
[0175][0176]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-1化合物(4.1g,10.6mmol)(式ⅲb化合物中ar为苯甲基),二氧六环(75ml),然后加热至内温70℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用二氧六环(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.5g(理论值的98.9%),de值99.6%。
[0177]
实施例14式ⅳc-1的制备
[0178][0179]
在室温下(约20℃),向反应瓶中加入式ⅱ化合物(5.0g,10.6mmol)、式ⅲb-1化合物(4.1g,10.6mmol)(式ⅲb化合物中ar为苯甲基),2-丁酮(75ml),然后加热至内温70℃溶清,搅拌3h。随后,将混合物在5h内冷却至20℃,然后在该温度下搅拌15h,过滤,用2-丁酮(2.5ml)漂洗固体,45℃真空干燥,得到白色固体4.5g(理论值的98.9%),de值99.0%。
[0180]
实施例15式ⅰ化合物的制备
[0181][0182]
在室温下(约20℃),向反应瓶中加入式ⅳa-1的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.7g,收率98.8%,hplc纯度99.7%,ee值99.6%。ms:m/z=470[m 1]
。
[0183]
实施例16式ⅰ化合物的制备
[0184][0185]
在室温下(约20℃),向反应瓶中加入式ⅳa-2的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.7g,收率95.1%,hplc纯度99.8%,ee值99.5%。ms:m/z=470[m 1]
。
[0186]
实施例17式ⅰ化合物的制备
[0187][0188]
在室温下(约20℃),向反应瓶中加入式ⅳc-1的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.6g,收率94.9%,hplc纯度99.6%,ee值98.6%。ms:m/z=470[m 1]
。
[0189]
实施例18式ⅰ化合物的制备
[0190][0191]
在室温下(约20℃),向反应瓶中加入式ⅳc-2的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.5g,收率97.7%,hplc纯度99.7%,ee值99.7%。ms:m/z=470[m 1]
。
[0192]
实施例19式ⅰ化合物的制备
[0193][0194]
在室温下(约20℃),向反应瓶中加入式ⅳc-3的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.5g,收率95.4%,hplc纯度99.5%,ee值99.8%。ms:m/z=470[m 1]
。
[0195]
实施例20式ⅰ化合物的制备
[0196][0197]
在室温下(约20℃),向反应瓶中加入式ⅳc-4的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.6g,收率98.5%,hplc纯度99.5%,ee值99.6%。ms:m/z=470[m 1]
。
[0198]
实施例21式ⅰ化合物的制备
[0199][0200]
在室温下(约20℃),向反应瓶中加入式ⅳc-5的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.3g,收率96.6%,hplc纯度99.6%,ee值99.5%。ms:m/z=470[m 1]
。
[0201]
实施例22式ⅰ化合物的制备
[0202][0203]
在室温下(约20℃),向反应瓶中加入式ⅳc-6的拆分盐(5.0g)、水(50ml),搅拌30分钟,然后滴加磷酸钠水溶液(100g磷酸钠溶于1000ml水中)调节ph至7~7.5,继续搅拌3h。过滤,用20ml水漂洗固体,45℃真空干燥,得到白色固体2.5g,收率93.6%,hplc纯度99.7%,ee值99.7%。ms:m/z=470[m 1]
。
[0204]
实施例23式
ⅰ‑
1化合物的制备
[0205][0206]
在室温下(约20℃),向反应瓶中加入式ⅰ化合物(5.0g,10.6mmol),四氢呋喃(50ml),10%钯炭(0.5g),氮气置换三次然后氢气置换三次,继续在室温条件下搅拌3h,过滤钯碳,浓缩有机相,得到白色固体3.9g,收率98.0%,ee值99.9%。hplc纯度99.7%。ms:m/z=380[m 1]
。
[0207]
实施例24非奈利酮的制备
[0208][0209]
在室温下(约20℃),向反应瓶中加入式
ⅰ‑
1化合物(1.6g,4.22mmol)、1,1-羰基二咪唑(1.0g,5.91mmol)、四氢呋喃(8ml),并在室温条件下加入4-二甲氨基吡啶(51mg,0.42mmol)。将混合物在室温条件下搅拌1小时,然后加热至50℃持续搅拌2.5小时。将六甲基二硅氮烷(3.0g,18.42mmol)加入至该溶液中,并在回流状态下搅拌22小时。再加入四氢呋喃(1.8ml),并将混合物冷却至5℃。在3小时内加入1.2ml四氢呋喃和1.0ml水的混合物,控制内温在5至20℃。随后将混合物在回流状态下搅拌1小时,缓慢降温至0℃,并在该温度下搅拌1小时,过滤,并用3ml水漂洗两次,55℃真空干燥,得到类白色固体1.5g,收率94.0%,hplc纯度99.9%,ee值100.0%。1h nmr(400mhz,[d6]dmso):δ=1.05(t,3h),2.12(s,3h),2.18(s,3h),3.82(s,3h),3.96
–
4.06(m,2h),5.37(s,1h),6.49
–
6.86(2h),7.15(d,1h),7.28(d,1h),7.37(d,1h),7.55(s,1h),7.68(s,1h)。ms:m/z=379[m 1]
。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。