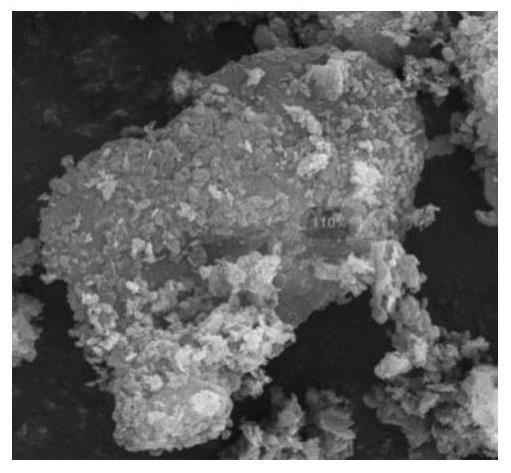
1.本发明属于超级电容器电极材料制备技术领域,具体涉及一种片状镍钴双金属有机框架晶体材料及其制备方法。
背景技术:
2.金属有机框架(mof)材料因其表面积高、活性位点丰富、结构多样且易于调控,在超级电容器电极材料领域引起广泛关注[wang y,et al.acs applied energy materials,2019,2(3)]。研究发现,片状结构的金属有机框架材料能够增大材料的比表面积、扩大与电解液的接触面积,加速电解液离子的传输。双金属离子之间的协同作用相较单金属有更强的电子储存能力[chen c,et al.2018,47,5639-5645],表现出更为优异的电化学性能。
[0003]
现有的金属有机框架材料合成路径常用水热法或溶剂热法,得益于水热反应高温高压的特点,得到的金属有机框架材料具有均匀的形貌特征。gao等采用水热法合成了蒲公英形状的nico-mof,钴离子部分取代了ni-mof结构中的镍离子,不仅使结晶度降低而且使mofs的结构从杨梅状转变为蒲公英状。较低的结晶度增强了导电性,蒲公英状球表面上分散的纳米棒有利于离子传输,这些都增强了电容性能[gao s,et al.journal of colloid and interface science,2018,531:83-90]。zhang等合成了片状镍钴双金属有机框架,所制备的片状nico-mof电极具有高比电容、良好的倍率性能以及长期循环稳定性[zhang x,et al.journal of materials science:materials in electronics,2020,27(4)];habib gholipour-ranjbar等利用吡嗪溶液兼具溶剂及配体作用,通过水热法合成了镍钴基双金属有机框架,所制备的双金属有机框架具有良好的速率性能及循环保持率。与ni-mof相比,nico-mof具有较低的电荷转移电阻和离子扩散势垒[gholipour-ranjbar h et al.new journal of chemistry,2016:10.1039.c6nj01449f]。
[0004]
目前研究多为由单配体制备的镍钴双金属有机框架,鲜有关于双配体结合的镍钴双金属有机框架的报道。且大部分所制备的双金属有机框架材料及其复合材料多为粉末状态,应用时需要使用粘结剂制备电极材料,大大降低了电极体系是导电性。
技术实现要素:
[0005]
为了解决背景技术中的问题,本发明提供了一种片状镍钴双金属有机框架晶体材料及其制备方法。该制备方法充分利用双金属镍离子、钴离子之间以及与双配体的双向协同作用,通过水热反应的高温高压作用及调控各项反应参数可以在活性碳布表面直接生长片状结构的镍钴双金属有机框架晶体,所获得的片状结构的复合材料具有低密度、高的比表面积及良好的导电性,在超级电容器电极材料的应用中占有很好的优势。
[0006]
本发明采用的技术方案如下:
[0007]
一、一种片状镍钴双金属有机框架晶体材料
[0008]
以二甲基咪唑和对苯二甲酸作为混合有机配体,六水合硝酸镍、六水合硝酸钴为反应物,通过水热反应获得负载在活性碳布表面的片状镍钴双金属有机框架晶体;镍钴双
金属有机框架晶体呈现均匀的片状结构,其平均厚度为0.15-0.20μm。
[0009]
二、一种片状镍钴双金属有机框架晶体材料的制备方法
[0010]
包括以下步骤:
[0011]
1)将六水合硝酸镍、六水合硝酸钴溶解于二甲基咪唑溶剂中,搅拌均匀得到混合溶液a;
[0012]
2)在混合溶液a中加入对苯二甲酸及聚乙烯吡咯烷酮,继续搅拌得到混合溶液b;聚乙烯吡咯烷酮的用量为1.5g;
[0013]
3)将处理后的活性碳布浸泡在混合溶液b中超声1h;
[0014]
4)将步骤3)的溶液连同活性碳布一起在100ml反应釜中进行水热反应;
[0015]
5)将步骤4)反应结束后的反应釜自然冷却至常温,经无水乙醇、去离子水反复清洗三次,干燥后获得均匀生长在活性碳布表面的片状镍钴双金属有机框架晶体。
[0016]
所述步骤1)中:
[0017]
六水合硝酸镍与六水合硝酸钴的摩尔质量比为1:1;
[0018]
二甲基咪唑溶剂的摩尔浓度为0.2mol/l,用量为30ml。
[0019]
混合溶液a中的镍离子和钴离子的总摩尔浓度为0.06mol/l-0.30mol/l。
[0020]
所述步骤2)中的对苯二甲酸与步骤1)中的二甲基咪唑的体积比为1:1。
[0021]
金属盐离子总物质的量与混合配体的总摩尔比为1:1-5:1;所述金属盐离子为镍离子和钴离子;所述混合配体为对苯二甲酸和二甲基咪唑。
[0022]
所述步骤3)中,活性碳的处理方式为:用丙酮、乙醇处理活性碳布,以去除表面油脂。
[0023]
所述步骤3)中,活性碳布为1cm
×
2cm亲水型商用碳布,厚度为0.36mm,纵向电阻小于0.12
×
10-2
ω,由预氧化的聚丙烯腈纤维织物经炭化而成,具有良好的导电性、柔韧性和大比表面积等特点。
[0024]
所述步骤4)中,水热反应的反应温度为150℃-180℃,反应时间在12h-18h。
[0025]
所述步骤5)中,干燥的温度为80℃-100℃,干燥时间为12h-24h。
[0026]
本发明采用温和、低能耗、快捷、安全的水热合成方法,选择可溶性的六水合硝酸镍、六水合硝酸钴为反应物,以二甲基咪唑和对苯二甲酸作为混合有机配体,通过调节反应参数得到在活性碳布表面自支撑生长的镍钴双金属有机框架晶体材料。
[0027]
本发明的有益效果是:
[0028]
1)本发明所制备的波浪片状结构晶体尺寸分布均匀、结晶度良好,化学成分均一,在扫描窗口为0-0.6v、电流密度为2ma/cm2时,所制得的nico-mof复合电极材料的比电容达到了834f/g,具有较高的比容量以及倍率性能。
[0029]
2)本发明以羧酸类化合物对苯二甲酸和二甲基咪唑为混合有机配体:
[0030]
羧酸类化合物对苯二甲酸,又称p-苯二甲酸,两个羧基分别与苯环中相对的两个碳原子相连接而成的二元芳香羧酸,具有以下两个作用:一是作为第一有机配体与金属离子反应,对苯二甲酸含有两个对角羧酸基团,羧酸基团可以与镍钴离子形成均匀网络,金属离子与羧酸基团的均匀连接导致更长的链长,并提高表面积和孔隙率;二是与第二配体二甲基咪唑协同反应形成混合网络结构,对苯二甲酸中的羧酸基团及含碳骨架能够与二甲基咪唑中的含氮活性位点发生桥联,能够有效增加有机配体和金属离子的配位位点,从而增
加了氧化还原活性位点。
[0031]
二甲基咪唑具有以下三重作用:一是作为反应溶剂,促使镍和钴的金属盐与有机配体充分溶解;二是作为第二有机配体与金属离子反应,二甲基咪唑分子有两个功能(氮)位点可以与金属离子配位形成网络;三是与第一有机配体对苯二甲酸协同反应,对苯二甲酸中的羧酸基团以及二甲基咪唑中的功能氮基团可以与金属离子形成混合配位从而引导原子微观排列,因此可以通过调节对苯二甲酸及二甲基咪唑的摩尔比形成均匀的网络结构,配位得到的双金属有机框架晶体受到双配体协同影响,具有可调控的分子形态和结构。本发明中双配体的协同作用形成网络结构,诱导生成二维片状形貌。片状结构能够增大电极材料与电解质溶液接触面积、加速离子的传输从而提升性能,制备方法简单易操作,有望应用在新型电极材料、催化、传感等领域。
[0032]
3)本发明以活性碳布为基底材料,活性碳布属于惰性材料,相比较其他导电衬底例如泡沫镍等更具柔韧性,且不腐蚀、不锈蚀、也不易被有害介质侵蚀,耐久性好,在电流传导上更具有优势。本发明充分利用活性碳布纤维表面的多孔结构,可增大与电解液接触面积,增大接触面的压力,可以减少其接触电阻等特殊功能,为金属有机框架晶体在碳布表面结晶提供良好条件,同时利用活性碳布优异的导电性能改善金属有机框架材料低导电性的缺点。
[0033]
4)本发明以聚乙烯吡咯烷酮作为表面活性剂,由于nico-mof纳米颗粒粒度小、表面能高易团聚,加入聚乙烯吡咯烷酮以后包裹在活性碳布表面可以阻碍团聚也能控制材料形状,同时聚乙烯吡咯烷酮起到分散离子及稳定的作用。
附图说明
[0034]
图1是实施例1所得产物的xrd图谱。
[0035]
图2是实施例1所得产物的电镜照片。
[0036]
图3是实施例2所得产物的电镜照片。
[0037]
图4是实施例3所得产物的电镜照片。
[0038]
图5是对比例1所得产物的电镜照片。
[0039]
图6是对比例2所得产物的电镜照片。
[0040]
图7是对比例3所得产物的电镜照片。
[0041]
图8是对比例4所得产物的电镜照片。
[0042]
图9是对比例5所得产物的电镜照片。
具体实施方式
[0043]
本发明通过使用双配体制备在活性碳布表面自支撑生长的双金属有机框架晶体材料。活性碳布是一种常见的导电基地,具有集流体材料功能,且具有柔韧性和大比表面积等特点,在其表面自支撑生成金属有机框架可以构筑复合电极材料,有效提高电极材料的电化学性能。双配体的协同作用、晶体材料片状形貌、自支撑结构设计及无粘结剂体系构建均有利于提高电化学性能。
[0044]
本发明制备的镍钴双金属有机框架晶体呈现均匀的片状结构,其平均厚度为0.15-0.20μm,片状结构增大了与电解液的接触面积,加快离子传输速率,从而提高性能。
[0045]
实施例1:
[0046]
将0.2910g六水硝酸钴、0.2910g六水硝酸镍和0.1500g对苯二甲酸溶于30ml二甲基咪唑溶液中。向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在150℃条件下反应12h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得负载在活性碳布上的片状镍钴双金属有机框架晶体。
[0047]
图1是实施例1产物的xrd图谱,该图谱与ccdc晶体库中模拟的nico-mof(no.638866)相匹配,证明在活性碳布表面原位生长的即是镍钴双金属有机框架晶体,在2θ为20
°
到30
°
之间出现的衍射峰与石墨晶体(001)和(110)晶面对应,说明亲水型商用碳布为类石墨结构;图2是实施例1产物的电镜照片,在活性碳布表面形成均匀的片状结构,片状厚度为100-200nm。
[0048]
实施例2:
[0049]
将0.5810g六水硝酸钴、0.5810g六水硝酸镍和0.1500g对苯二甲酸溶于30ml二甲基咪唑溶液中。向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在150℃条件下反应16h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得负载在活性碳布上的片状镍钴双金属有机框架晶体。
[0050]
图3是实施例2产物的电镜照片,活性碳布表面形成均匀的片状结构,片状厚度为100-150nm。
[0051]
实施例3:
[0052]
将1.4530g六水硝酸钴、1.4530g六水硝酸镍和0.1500g对苯二甲酸溶于30ml二甲基咪唑溶液中。向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在180℃条件下反应18h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得负载在活性碳布上的片状镍钴双金属有机框架晶体。
[0053]
图4是实施例3产物的电镜照片,活性碳布表面形成均匀的片状结构,片状厚度为200-300nm。
[0054]
对比例1:
[0055]
将0.5810六水硝酸钴、0.9880g六水硝酸镍和0.1500g对苯二甲酸溶于30ml二甲基咪唑溶液中。向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在180℃条件下反应18h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得最终产物。
[0056]
图5是对比例1产物的电镜照片,活性碳布表面未形成片状结构,而是无定型状。
[0057]
对比例2:
[0058]
将0.5810六水硝酸钴、0.5810g六水硝酸镍和0.5000g对苯二甲酸溶于30ml二甲基咪唑溶液中。向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在150℃条件下反应12h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得最终产物。
[0059]
图6是对比例2产物的电镜照片,活性碳布表面未形成片状结构,而是无定型状。
[0060]
对比例3:
[0061]
将0.5810六水硝酸钴、0.581g六水硝酸镍和0.15g对苯二甲酸溶于30ml二甲基咪唑溶液中,向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在120℃条件下反应8h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得最终产物。
[0062]
图7是对比例3产物的电镜照片,活性碳布表面未形成片状结构,而是无定型状。
[0063]
对比例4:
[0064]
将0.5810六水硝酸钴、0.581g六水硝酸镍和0.15g对苯二甲酸溶于30ml二甲基咪唑溶液中,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在150℃条件下反应12h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得最终产物。
[0065]
图8是对比例4产物的电镜照片,活性碳布表面未形成片状结构,而是无定型状。
[0066]
对比例5:
[0067]
将0.5810六水硝酸钴、0.581g六水硝酸镍溶于30ml二甲基咪唑溶液中,向混合溶液中加入1.5000g聚乙烯吡咯烷酮,磁力搅拌30min得到均匀溶液。随后将处理后的活性碳布浸入混合溶液中超声1h,将混合均匀溶液连同活性碳布倒入100ml反应釜中,在150℃条件下反应12h,反应结束后待反应釜自然冷却至常温,将得到的活性碳布用无水乙醇、去离子水反复离心清洗三次,除去表面吸附离子、附着物等杂质,并在80℃干燥12h,获得最终产物。
[0068]
图9是对比例5产物的电镜照片,活性碳布表面未形成片状结构,而是无定型状。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。