1.本发明属于液态有机化合物储氢领域及催化材料技术领域,具体涉及一种氢化芳香族化合物的低温脱氢方法。
背景技术:
2.氢气在国民经济各行各业中具有广泛的用途。氢气既可以作为石油化工原料,又是一种清洁、高效、安全、可持续的新能源,在技术上能够解决人类面临的能源与环境两大方面的问题,因而国际上认为氢能将是最理想的能源,也是人类长远的战略能源。
3.氢能相关技术包括氢气的大规模制取、储运和高效利用。氢气的常温常压储存和输送是制约其大范围应用的关键瓶颈。开发出高效的液态储氢材料,实现氢气的可逆存储和释放,将成为解决整个氢气产业链的重要环节。
4.液体储氢材料在富产氢之地加氢得到同样为液态的氢化物,经过脱氢释放出氢气。脱氢过程是强吸热的非均相反应,且反应过程可逆,从动力学和热力学两方面考虑,必须在低压高温的条件下进行,然而高温容易导致催化剂积炭、裂解反应的发生,降低催化剂的活性和主要产物的选择性,进而影响脱氢反应过程的稳定性和催化剂的寿命。因此影响有机液体氢化物储氢技术大规模应用的关键是开发低温高选择性的脱氢催化剂。
5.通常使用的脱氢催化剂为负载型金属催化剂,活性组分为pt、pd、rh、ni、co等。也有通过添加第二金属组分如ni、mo、w、re、rh、pd、ir、sn等,可进一步提高催化剂的脱氢活性。载体一般是氧化铝或者氧化铝的改性组分。
6.大多数的商售活性氧化铝表面羟基过多、酸性过强。使用这类氧化铝为载体制备脱氢催化剂,在反应过程中催化剂表面易于积碳,进而导致迅速失活。此外活性组分的前驱体中往往含有呈酸性的氯离子,活性组分浸渍过程中采取的竞争吸附剂也经常采用盐酸溶液,均会引入氯离子,增加催化剂的酸性,使得催化剂表面酸强度进一步增强。催化剂中酸性要适中,酸性过强,会发生异构化甚至加氢裂解作用,生成副产物,降低脱氢选择性。酸性过弱,催化剂的活性会降低,影响脱氢转化率。
7.因此,针对催化剂的使用需求,需要对催化剂进行适当的降酸处理。有的直接采用氧化铝之外的非酸性物质做载体,如活性炭、分子筛等。有的采用非氯离子型活性组分的前驱体,有的采用洗氯的方式,将催化剂中残留的氯离子洗掉。有的采用将催化剂中引入碱性或碱土金属弱化载体表面的酸性。有的在碱性环境下对载体进行改性,并且在碱性环境下负载活性组分。
8.cn109701610a涉及到一种改性脱氢催化剂、制备方法及其用途,所涉及的催化剂包括两类活性组分,组分(a)为铂系金属的一种或多种,组分(b)为in、cs、ga、ge、sr中的一种或多种,载体为氧化铝、分子筛和氧化硅中的一种或多种,涉及到的改性是分子筛载体经过氮改性。涉及到的有机液体储氢材料选自甲基环己烷、环己烷、四氢萘、十氢萘、全氢氮乙基咔唑和全氢咔唑中的一种或多种。实施案例显示,涉及到的载体为分子筛,合成的催化剂在320℃下,对于环己烷、四氢萘、十氢萘、甲基环己烷而言,初始转化率最高为82%,反应
100h后,转化率下降为79%,结焦率达到0.4%。
9.cn105582917b公开一种贵金属脱氢催化剂的脱氯方法,具体是在催化剂床层通入质量浓度为1%~10%氨水溶液进行水热脱氯处理,水热脱氯处理结束后,通入去离子水进行水热脱氨处理,得到脱氯后的贵金属脱氢催化剂。该方法可将贵金属脱氢催化剂中的cl-脱除至0.15wt%以下,避免了高温下pt颗粒的聚集长大,降低了能耗。
10.cn110252422a公开一种催化剂中氯离子的洗涤方法,该方法用含有铵盐、或铵盐和还原剂的洗涤液洗涤负载活性组分后的半成品催化剂3~20次,烘干后焙烧,即可得到氯离子含量在10ppm以下的催化剂。
11.cn112452340a、cn112371193a、cn112316977a等多项专利公开了去离子水洗氯离子的方法,均是反复洗涤,将溶液洗涤至中性,即将酸性物质全部洗掉。
12.cn111889094a提供了一种有机储氢化合物脱氢制氢气催化剂,包括贵金属活性组分、氧化铝和改性金属氧化物,所述的改性金属氧化物为钛氧化物和/或锆氧化物,即载体为氧化铝和改性金属氧化物的混合物。该脱氢催化剂具有更好的脱氢活性和选择性。实施案例中显示,用于甲基环己烷脱氢,在350℃下,压力为1mpa时,脱氢转化率最高可达到77%,选择性为95.8%。
13.cn110882703a公开了一种含碱土金属的环烷烃脱氢催化剂及其制备方法,以pt为活性金属组分,以sn为助剂组分,载体为含碱土金属、硫和钛的氧化铝载体。在载体原位制备载体的过程中所形成的碱土金属-ti-al骨架结构能够显著改善载体表面氧化铝酸性单一的特点,显著降低氧化铝载体的酸性,提高催化剂的抗积炭性能,提高催化剂的高温活性和稳定性。优选用于环己烷脱氢制苯或甲基环己烷脱氢制甲苯。实施案例显示,其将催化剂用于环己烷脱氢,反应温度为430℃,压力1.0mpa,空速为2h-1
时,转化率最高为92%,选择性为84%。
14.美国专利us3531543公开了用由铂、锡及中性金属氧化物载体组成的催化剂,进行烃类脱氢。优选的载体是氧化物物料,其固有酸性已基本上被碱金属或碱土金属组份中和掉。此外,随碱金属含量的增加,这些氧化铝的酸性也相应地减少。该专利的载体最好是一种非酸性的加氧化锂的氧化铝。该专利的催化剂最好由不含囟素的化合物制成。但只要从最后的催化剂复合物中可有效地除去残留的囟素,含囟素的化合物也可以用来制造催化剂。
15.美国专利us3745112公开了一种用于烃类重整的催化剂,它由铂族组份、锡组份及囟素组份及多孔载体物质构成。该专利还公开了铂-锡-碱金属或碱土金属的复合物是一种特别有效的烃类脱氢催化剂。在该专利的脱氢催化剂复合物中加有碱金属或碱土金属组份,所以囟素的量如果没有完全排除,也被减至极小,以便使氧化铝与囟素组份的酸性作用减至极小或中和掉;该酸性作用会促进工业脱氢过程所不希望有的烃类裂解和异构化副反应。
16.美国专利us3892657公开了当铟与铂的原子比是大约0.1:1到大约1:1时铟是含铂族催化剂的良好助剂。该专利还公开了将选自锗、锡、铅的一个ⅳa族组份加入到可以用于重整应用的含铟酸性催化剂中。从而这种酸性催化剂由铂族组份、ⅳa族组份、铟组份、囟素组份和多孔载体物质构成。对于重整方面的应用,酸性催化剂含有高达大约3.5%(重量)的囟素,而对于异构化及裂解方面的应用,酸性催化剂含有高达大约10%(重量)的囟素。该专
利的脱氢催化剂中,尽管加有碱金属或碱土金属组份,囟素含量被保持于可能的最低值,大约0.1%(重量)。
17.美国专利us3909451公开了制作一种含铂组份、锡组份及一种碱金属或碱土金属组份的脱氢催化剂的新方法。该专利在实例
ⅴ
中公开了一种铂、锡及钾的组合物,它含有少于0.2%(重量)的化合态氯。
18.美国专利us4329258和us4363721公开了一种用耐熔氧化物-矿物作载体由铂族金属、锡、一种碱金属或碱土金属和化合态囟素组成的催化剂。这些专利的催化剂中碱金属或碱土金属和铂族金属的原子比是从0.2到10。专利权人发现,把百万分之几的碱金属或碱土金属组份加到含有铂族金属、锡及囟素的催化剂中有助于增加重整过程中c 5的产率。
19.英国专利1499297公开了一种用氧化铝作载体由铂、及镓铟铊元素中的至少一个、以及碱金属(特别是锂和钾)组成的脱氢催化剂。该专利的催化剂也含有数量从0.01到0.1%(重量)的囟素。囟素含量被特意地降低到这样低的重量百分数范围内是为了增加催化剂的选择性和稳定性。
20.专利cn 111686718 a在碱溶液中对载体进行处理,并在碱性环境下负载活性组分,以增大浸渍液中金属负载化合物的空间位阻和金属负载速率,防止金属粒子团聚,提高催化剂活性组分的分散性和均一性。结果显示,在430~450℃下转化率可达到99.5%,选择性达到99%以上。
21.综上所述,涉及到脱氢的化合物主要包括低链烷烃(如丙烷、丁烷)和环烷烃及含杂原子芳烃的氢化物。为了降低催化剂表面酸性,提高脱氢催化剂的稳定性,研究者们一般采取两种措施,一是将氯完全脱除或者保持尽可能低的水平,按元素为基础计算时一般少于0.1%(重量),并总是少于0.2%(重量)。二是载体制备过程中掺杂碱金属或碱土金属的方式,对催化剂表面的酸起到削弱作用。
22.在现有技术中,上面所知的脱氢催化剂由铂族组份、ⅳa族组份以及一种碱金属或碱土金属组份构成,其中碱金属或碱土金属组份与铂族组份的原子比大于10,而囟素组份已被完全排除或者保持在可能的最低水平。
技术实现要素:
23.本发明的目的在于克服现有的脱氢催化剂转化率和选择性均不高的缺陷,尤其是在低温条件下的转化率低。本发明提供一种氢化芳香族化合物的低温脱氢方法,该催化剂的反应活性高,对氢化芳香族化合物的脱氢转化率高,选择性能达到100%。
24.本发明采取的技术方案如下:
25.本发明提供了一种氢化芳香族化合物的低温脱氢方法,该氢化芳香族化合物的低温脱氢方法,包括将富含氢化芳香族化合物的原料及脱氢催化剂加入到脱氢反应器中,在温度:240~420℃,压力:0.01~2mpa,质量空速:1~5h-1
,氢油摩尔比为0~1.0的条件下,进行脱氢反应。该脱氢催化剂为非硫化型催化剂,包括活性组分、金属助剂、氯离子和载体组成。活性组分至少含有第viii族的贵金属组分,占脱氢催化剂总重量的0.1~3%。氯离子占脱氢催化剂总重量的0.3~1.2%。金属助剂至少含有碱金属和/或碱土金属,占脱氢催化剂总重量的0.3~0.9%,金属助剂与氯原子摩尔比为0~1.5。载体主要为γ-al2o3。
26.作为优选地,活性组分占低温脱氢催化剂总重量的0.1~0.6%;氯离子占低温脱
氢催化剂总重量的0.35~0.9%;金属助剂与氯原子摩尔比为0.4~1.2。
27.本发明的活性组分包括pt、pd、rh中的至少一种物质。
28.作为优选地,活性组分为pt。
29.载体主要为γ-al2o3,载体中γ-al2o3的含量》50wt%,最好是100wt%,载体中还可以包含gro2,ceo2、分子筛、活性炭等。
30.本发明并不特别限定载体的形状,可以是球形、条形、柱形和三叶草形中的至少一种。
31.不同原料所需要的载体形貌结构不同,本发明不作特别限定,对于本发明通常比表面积为180~240m2/g,孔容为0.5~2.0ml/g,孔径为2~20nm。
32.上述技术方案中,富含氢化芳香族化合物的原料是指富含芳香族化合物经过加氢后得到完全加氢或者部分加氢的产物。
33.上述技术方案中,芳香族化合物包括1~3环芳烃和1~3环含n杂原子的杂环芳烃。1~3环芳烃可以含有0~3个侧链,侧链长度为1~3个碳数。
34.本发明不限定脱氢反应形式,但本技术方案中采用固定床反应形式。
35.脱氢反应条件优选为反应温度:260~320℃,反应压力:0.01~0.5mpa,质量空速:1~3h-1
,氢油摩尔比为0~0.6。
36.本发明不特别限定脱氢催化剂的制备方法,但是优选采用如下制备过程:
37.脱氢催化剂的制备方法可以包含以下步骤:
38.s1:配置含活性组分的金属盐溶液,调节ph值为0~3,形成含氯的活性组分浸渍液;
39.s2:将浸渍液和载体混合,浸渍3~6h,去除残余溶液后在120~200℃下干燥2~6h,移入管式炉,在400~600℃下焙烧4~8小时,降温后得到脱氢催化剂前驱体-1;
40.s3:配置含金属助剂的金属盐溶液,形成金属助剂浸渍液,与脱氢催化剂前驱体-1混合,浸渍3~6h,去除残余溶液后在120~200℃下干燥2~6h,移入管式炉,在400~600℃下焙烧3~6小时得到脱氢催化剂前驱体-2;
41.s4:将脱氢催化剂前驱体-2在300~600℃下的氢气气氛中还原4~8h,得到脱氢催化剂。
42.本发明的脱氢催化剂还可以按照以下步骤进行制备:
43.s1:配置含活性组分和金属助剂的金属盐溶液,调节ph值为0~3,形成含氯的活性组分和金属助剂浸渍液;
44.s2:将浸渍液和载体混合,浸渍3~6h,去除残余溶液后在120~200℃下干燥2~6h,移入管式炉,在400~600℃下焙烧4~8小时,降温后得到脱氢催化剂前驱体;
45.s3:将脱氢催化剂前驱体在300~600℃下的氢气气氛中还原4~8h,得到脱氢催化剂;
46.本发明的脱氢催化剂还可以按照以下步骤进行制备:
47.s1:配置含金属助剂的金属盐溶液,形成金属助剂浸渍液,与载体混合,浸渍3~6h,去除残余溶液后在120~200℃下干燥2~6h,移入管式炉,在400~600℃下焙烧3~6小时得到脱氢催化剂前驱体-1;
48.s2:配置含活性组分的金属盐溶液,调节ph值为0~3,形成含氯的活性组分浸渍
液;
49.s3:将活性组分浸渍液和脱氢催化剂前驱体-1混合,浸渍3~6h,去除残余溶液后在120~200℃下干燥2~6h,移入管式炉,在400~600℃下焙烧4~8小时,降温后得到脱氢催化剂前驱体-2;
50.s4:将脱氢催化剂前驱体-2在300~600℃下的氢气气氛中还原4~8h,得到脱氢催化剂。
51.上述技术方案中,所述的活性组分包括pt、pd、rh中的至少一种物质,优选pt,活性组分的金属盐溶液可以是氯酸盐溶液、硝酸盐溶液、硫酸盐溶液的中一种或多种。
52.上述技术方案中,所述的金属助剂对应的金属盐为氯化物、氢氧化物、硝酸盐、硫酸盐、柠檬酸盐和草酸盐中的至少一种。
53.上述技术方案中,所述的含氯的浸渍液中的氯可以来源于活性组分的氯酸盐溶液,也可以来源于浸渍液中所加入的含氯的酸性组分,也可以来源于金属助剂的金属盐。
54.本发明浸渍后的载体不经过洗涤,在通入去离子水的流通的空气氛围中完成。空气携带去离子水的方式不限,可以使空气通过常温去离子水,沸腾的热水或者配置成一定浓度的hcl溶液,焙烧通过的空气气流空速为16~20min-1
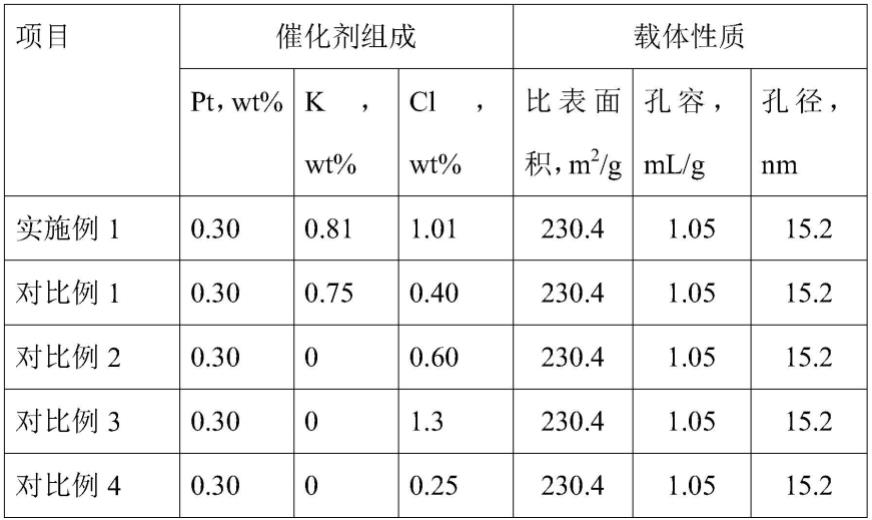
。
55.本发明不强调焙烧过程的升温模式,但本技术方案中优选程序升温,升温速率为2~5℃/min。
56.本发明通过研究发现,在催化剂制备过程中,通过采取降低催化剂的氯含量,能够有效降低催化剂的表面酸性,提高脱氢转化率以及选择性,但是需要控制洗氯条件,保证氯离子含量适中处于特定的含量,太低或者太高,转化率提高均有限,在脱氯基础上再引入金属金属助剂,会进一步提高低温转化率。在将氯洗涤至一合理区间的基础上,再在催化剂上负载上一定量的碱金属或碱土金属,在保持选择性的同时,可以进一步显著提高催化剂的低温活性,使得化合物在各个温度下的转化率显著提高。
57.与现有技术相比,本发明具有以下优点:
58.(1)本发明所述脱氢催化剂组成简单,条件易于控制,产品重复性好,通过控制催化剂氯离子含量结合添加金属助剂改性,从两个层面改善了催化剂的表面酸性;
59.(2)脱氢反应原料来源广,可在工业上大规模制备;
60.(3)反应形式可以是固定床,成熟可靠;
61.(4)脱氢催化剂用于芳香族化合物加氢产物的脱氢时,反应条件缓和,转化率和选择性高,表现出优异的低温脱氢活性。
具体实施方式
62.下面通过实施例对本发明进行具体描述。有必要在此指出的是以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员可以根据上述本发明内容对本发明作出一些非本质的改进和调整。
63.实施例1
64.(1)催化剂
65.载体采用球形剂,γ-al2o3含量98wt%,比表面积为230.4m2/g,孔容为1.05ml/g,孔径为15.2nm。pt含量0.3wt%,cl含量为1.01wt%,k含量0.81wt%,k/cl摩尔比为0.73。
66.(2)催化剂制备
67.依次将4ml浓度为15mg/ml的氯铂酸盐溶液,8ml浓度为80mg/ml的盐酸溶液加入三角瓶,再加入28ml去离子水,调节ph值为1,配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,在空气携带去离子水进入管式炉,采用程序升温,升温速率为2℃/min,升温到500℃下焙烧4h,冷却后取出得到催化剂前驱体-1。配置36ml钾离子浓度为13.32mg/ml的硝酸钾溶液,加入20g焙烧后的催化剂前驱体-1中,浸渍4h后过滤除去多余的溶液,在120℃下干燥2h后再次转移到管式炉中,在500℃的空气气氛中焙烧4h后,得到催化剂前驱体-2。催化剂前驱体-2在500℃,氢气氛围下还原4h,得到催化剂a,其组成数据见表1。
68.(3)脱氢反应方法
69.脱氢在固定床反应体系进行,在固定床反应器内装填10g制备的催化剂a,设置反应装置压力为0.1mpa,质量空速为2h-1
,氢油摩尔比为0.5,进原料后开始升温,从260℃开始,每隔6h取样后,提高温度20℃,直到400℃。产物采用gc-ms分析产物组成,根据组成计算原料脱氢的转化率和生成目标芳烃-全脱氢产物的选择性。分别评价催化剂对苯、甲苯、萘、咔唑、n-乙基咔唑等氢化芳烃化合物的脱氢转化性能。对于参与评价的几种原料,从自身结构而言,除环己烷外,甲基环己烷反应难度最大,能反应出催化剂的反应性能以及工艺条件对转化率的影响,因此实施例中仅对甲基环己烷的脱氢转化率情况进行了说明。将甲基环己烷在不同温度下的脱氢效果列于表2。
70.对比例1
71.(1)催化剂
72.载体与实施例1相同。pt含量0.3wt%,cl含量为0.40wt%,k含量0.75wt%,k/cl摩尔比为1.71。
73.(2)催化剂制备
74.依次将4ml浓度为15mg/ml的氯铂酸盐溶液,5ml浓度为80mg/ml的盐酸溶液加入三角瓶,再加入27ml去离子水,调节ph值为1,配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,在空气携带去离子水进入管式炉,在500℃下焙烧6h,冷却后得到催化剂前驱体-1。配置36ml钾离子浓度为13.32mg/ml的硝酸钾溶液,倒入20g焙烧后的催化剂前驱体-1中,浸渍4h后过滤除去多余的溶液,在120℃下干燥2h后再次转移到管式炉中,在500℃的空气气氛中焙烧4h后,得到催化剂前驱体-2。催化剂前驱体-2在500℃,氢气氛围下还原4h,得到催化剂b,其组成数据见表1。
75.(3)脱氢反应方法
76.采用与实施例1相同的评价方法,结果见表2。
77.对比例2
78.(1)催化剂
79.载体与实施例1相同,pt含量0.3wt%,cl含量为0.60wt%,k含量0.0wt%,k/cl摩尔比为0。
80.(2)催化剂制备
81.依次将4ml浓度为15mg/ml的氯铂酸盐溶液,5ml浓度为80mg/ml的盐酸溶液加入三角瓶,再加入27ml去离子水,调节ph值为1,配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,在空气携带去离子水进入管式炉,在500℃下焙烧5h,得到催化剂前驱体-1,催化剂前驱体-1在500℃氢气氛围下还原4h,得到催化剂c,其组成数据见表1。
82.(3)脱氢反应方法
83.采用与实施例1相同的评价方法,评价结果见表2。
84.对比例3
85.(1)载体
86.载体与实施例1相同。pt含量0.3wt%,cl含量为1.3wt%,k含量0.0wt%,k/cl摩尔比为0。
87.(2)催化剂制备
88.依次将4ml浓度为15mg/ml的氯铂酸盐溶液,5ml浓度为80mg/ml的盐酸溶液加入三角瓶,再加入27ml去离子水,调节ph值为1,配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,在空气直接进入管式炉,在500℃下焙烧6h,得到催化剂前驱体-1,催化剂前驱体-1在500℃氢气氛围下,还原4h,得到催化剂d,其组成数据见表1。
89.(3)脱氢反应方法
90.采用与实施例1相同的评价方法,评价结果见表2。
91.对比例4
92.(1)催化剂
93.采用与实施例1相同的载体。pt含量0.3wt%,cl含量为0.25wt%,k含量0.0wt%,k/cl摩尔比为0。
94.(2)催化剂制备
95.依次将4ml浓度为15mg/ml的氯铂酸盐溶液,5ml浓度为80mg/ml的盐酸溶液加入三角瓶,再加入18ml去离子水,调节ph值为1。配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,空气携带水蒸汽进入管式炉,在500℃下焙烧6h,得到催化剂前驱体-1,催化剂前驱体-1在500℃氢气下氛围,还原4h,得到催化剂e,其组成数据见表1。
96.(3)脱氢反应方法
97.采用与实施例1相同的评价方法,评价结果见表2。
98.实施例2
99.(1)催化剂
100.载体与实施例1相同。pt含量0.52wt%,cl含量为0.67wt%,k含量0.43wt%,k/cl摩尔比为0.58。
101.(2)催化剂制备
102.依次将8ml浓度为15mg/ml的氯铂酸盐溶液,10ml浓度为80mg/ml的盐酸溶液加入三角瓶,加入0.414g硝酸钾,再加入27ml去离子水,调节ph值为0.5,配置成浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,空气携带水蒸气分子进入管式炉,在500℃下焙烧5h,冷却后得到催化剂前驱体,催化剂前驱体在500℃氢气氛围下还原4h,得到催化剂f,其组成数据见表1。
103.(3)脱氢反应方法
104.采用与实施例1相同的评价方法,评价结果见表2。
105.实施例3
106.(1)催化剂
107.载体与实施例1相同。pt含量0.3wt%,cl含量为0.50wt%,k含量0.40wt%,k/cl摩尔比为0.73。
108.(2)催化剂制备
109.将0.305g氯化钾加入36ml去离子水,配置成钾离子浸渍溶液。往浸渍液中加入20g载体,将三角瓶口用密封膜封住,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,在空气携带水蒸气进入管式炉,在500℃下焙烧4h,冷却后得到催化剂前驱体-1。将8ml浓度为15mg/ml的pt盐溶液和10ml浓度为80mg/ml的盐酸溶液加入三角瓶,调节ph值为0.5。加入18ml去离子水,配置成活性组分浸渍液,加入到催化剂前驱体-1中,放在摇床上振荡浸渍4h。取下三角瓶,将多余的浸渍液脱除后,放在烘箱中,在120℃下干燥2h后转移到管式炉中焙烧。焙烧所用的空气空速为16min-1
,空气携带水蒸气分子进入管式炉,程序升温,在500℃下焙烧4h,冷却后得到催化剂前驱体-2。催化剂前驱体-2在500℃的氢气氛围下还原4h,得到催化剂g,其组成数据见表1。
110.(3)脱氢反应方法
111.采用与实施例1相同的评价方法,评价结果见表2。
112.对比例5
113.(1)催化剂
114.与实施例1相同。
115.(2)催化剂制备
116.与实施例1相同
117.(3)脱氢反应方法
118.脱氢在固定床反应体系进行,反应器内装填10g制备的催化剂a,设置反应装置压力为2.5mpa,质量空速为2h-1
,氢油摩尔比为1.5,采用甲基环己烷为原料,进原料后开始升温,从260℃开始,每隔6h取样后,提高温度20℃,直到400℃。样品采用gc-ms分析产物组成,根据组成计算原料脱氢的转化率和生成目标芳烃甲苯的选择性。对甲基环己烷在不同温度下的脱氢效果列于表2。
119.实施例4
120.(1)催化剂
121.与实施例1相同。
122.(2)催化剂制备
123.与实施例1相同
124.(3)脱氢反应方法
125.与脱氢在固定床反应体系进行,反应器内装填10g制备的催化剂a,设置反应装置压力为0.1mpa,质量空速为0.5h-1
,氢油摩尔比为0,采用临氮气反应,反应原料为甲基环己烷,进原料后开始升温,在320℃下连续运转,每隔12h取样后。样品采用gc-ms分析产物组成,根据组成计算原料脱氢的转化率和生成目标芳烃甲苯的选择性。对甲基环己烷连续运转的脱氢效果列于表3。
126.实施例5
127.(1)催化剂
128.与实施例1中相同。
129.(2)催化剂制备
130.与实施例1相同
131.(3)脱氢反应方法
132.与脱氢在固定床反应体系进行,反应器内装填10g制备的催化剂a,设置反应装置压力为0.1mpa,质量空速为0.5h-1
,氢油摩尔比为0.5,反应原料为甲基环己烷,进原料后开始升温,在320℃下连续运转,每隔12h取样后。样品采用gc-ms分析产物组成,根据组成计算原料脱氢的转化率和生成目标芳烃甲苯的选择性。对甲基环己烷连续运转的脱氢效果列于表3。
133.对比实施例4和5可以得出,虽然非临氢条件下,反应初始活性更高,但长周期运转活性下降较快,因此临氢条件对于催化剂长周期运转更为有利。
134.经过对比发现,除了催化剂组成,工艺条件选取对脱氢转化率影响也很大,低压高温转化率更高。低温条件下,催化剂组成、反应条件对转化率影响很大,而在高温条件下,温度成为反应主导因素,催化剂组成、反应条件的影响很小。
135.表1催化剂的组成和性质
[0136][0137][0138]
表2催化剂对甲基环己烷的脱氢作用效果
[0139][0140]
表3临氢与否对脱氢体系长周期运转作用效果
[0141]
运转时间,h实施例4-非临氢实施例5-临氢12100.0099.8010080.9099.60
[0142]
应注意的是,以上实例仅用于说明本发明的技术方案而非对其进行限制。尽管参照所给出的实例对本发明进行了详细说明,但是本领域的普通技术人员可根据需要对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
