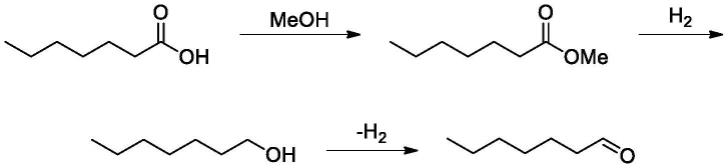
1.本发明属于精细化工领域,具体涉及一种以巴豆醛、烯丙醇为原料,制得正庚醛的方法。
背景技术:
2.庚醛是一种无色油状液体,与乙醇、乙醚等有机溶剂混溶,具有清鲜的果子香味,可以作为香料使用,用于配制柑橘类、蔬菜类和瓜果类香精;庚醛也是一种很重要的中间体,可以合成庚醇和庚酸酯香料,庚醛和苯甲醛缩合可以制备α-戊基肉桂醛,庚醛和原甲酸三乙酯反应可以得到庚醛二乙缩醛,这些产品都是香精香料产品。尤其是α-戊基肉桂醛,具有非常好的花香,是一种应用非常广泛的重要香料,可以用于茉莉、铃兰、紫丁香、风信子等香精,在皂用香精中用量高达2-10%。α-戊基肉桂醛还是我国规定允许使用的食用香精,可用于配制苹果、杏、桃、草莓味的食用香精,用量1-15mg/kg不等。
3.目前获得正庚醛的方法主要分为两类,一是正庚醇氧化法,通过氧化将醇转化为醛,不发生碳骨架的改变。该方法的起始原料是天然油脂,将天然油脂酯交换、然后精馏分离c7组分,高压加氢得到庚醇,最后氧化脱氢得到庚醛。该方法虽然路线较短,但是天然油脂组分复杂,分离纯化困难,而且天然油脂中c7含量较少,分离收率较低;生产过程中还涉及高压加氢以及脱氢反应,设备要求高、风险较大。
[0004][0005]
获得正庚醛的另一种方法是以蓖麻油为原料,先将蓖麻油皂化,得到蓖麻油酸甲酯,蓖麻油酸甲酯在高温下热裂解得到10-十一烯酸甲酯和正庚醛,其中正庚醛的收率20-30%不等,裂解得到的是10-十一烯酸甲酯和正庚醛的混合物,精馏纯化后得到正庚醛。该方法消耗大量的蓖麻油,裂解反应温度很高,收率也比较低。
[0006]
综上所述,目前合成正庚醛的主要方法是正庚醇氧化或者蓖麻油裂解,两种方法的原料都是天然来源,价格十分昂贵,要么反应路线长、要么反应条件苛刻,造成正庚醛的价格居高不下。因此,目前急需发展新型、高效的正庚醛合成路线,可以从价格低廉的起始原料出发,简洁、高效、条件温和的得到正庚醛。
技术实现要素:
[0007]
针对现有技术中存在的上述问题,本发明的目的在于提供一种以廉价易得的巴豆醛、烯丙醇为原料合成正庚醛的方法。
[0008]
为实现上述目的和达到上述技术效果,本发明采用如下技术方案:
[0009]
本发明提供一种由巴豆醛和烯丙醇合成正庚醛的方法,步骤包括:
[0010]
1)由巴豆醛和烯丙醇通过脱水缩合反应制得1,1-双(烯丙氧基)丁-2-烯中间体;
[0011]
2)1,1-双(烯丙氧基)丁-2-烯中间体通过裂解重排反应制得2,6-二庚烯醛;
[0012]
3)2,6-二庚烯醛通过选择性加氢反应制得正庚醛。
[0013]
本发明以巴豆醛为起始原料,经三步反应制得正庚醛的方法中,各步骤产物对应的结构式如下:
[0014][0015]
本发明中,步骤1)所述巴豆醛与烯丙醇的摩尔比为1:2-5,优选1:2-3。
[0016]
本发明中,步骤1)所述脱水缩合反应,温度为50-100℃,优选60-90℃,时间为1-24h,优选4-12h;
[0017]
优选地,所述脱水缩合反应在氮气氛围中进行,压力为10-101kpaa,优选50-101kpaa。
[0018]
本发明中,步骤1)所述脱水缩合反应可以在催化剂条件下反应,也可以在无催化剂条件下反应,出于经济性考虑,优选在催化剂条件下反应,为尽可能提高转化率和选择性,所述催化剂进一步优选为二叔丁基过氧化物;
[0019]
所述催化剂用量为巴豆醛质量的1.0-5.0wt%,优选2.0-4.0wt%。
[0020]
在一些具体示例中,在所述脱水缩合反应过程中,还优选添加铑金属化合物作为助剂,与催化剂协同作用,进一步提高1,1-双(烯丙氧基)丁-2-烯中间体的选择性;
[0021]
所述铑金属化合物选自铑的卤化物、铑与乙酰基化合物的配合物、铑与羰基化合物的配合物中的一种或多种;优选的,所述铑金属化合物选自rhcl3、rh(co)2acac、rh4(co)
12
、rh6(co)
16
中的一种或多种;
[0022]
所述铑金属化合物用量为巴豆醛质量的0.01-0.05wt%,优选0.02-0.04wt%。
[0023]
本发明步骤1)中添加铑金属化合物作为助剂能够显著提高1,1-双(烯丙氧基)丁-2-烯的选择性,由于原料巴豆醛和烯丙醇分子量小且含有双键,反应活泼性高,容易发生自身异构或聚合而降低反应选择性,通过添加铑金属化合物能够稳定其构型并可作为除氧剂除去体系中的氧,或者通过电子转移,将增长链自由基转为稳定分子,起到一定的阻聚作用,少量添加就可达到反应效果。
[0024]
本发明中,步骤1)所述脱水缩合反应为平衡反应,在一些具体示例中,还优选加入共沸溶剂,采用共沸带水方式进行,不断将水移出反应体系,促进缩合反应进行,待反应体系再无水移出后,代表反应缩合反应基本进行完全;所述共沸溶剂包括但不限于甲苯、正己烷、环己烷、乙腈、二甲苯等中的一种或多种;
[0025]
所述共沸溶剂用量为巴豆醛质量的10.0-50.0wt%,优选20.0-40.0wt%。
[0026]
本发明中,步骤1)所述脱水缩合反应完成后,还包括分离、脱溶剂等后处理过程,如降温,反应液分相(上层为1,1-双(烯丙氧基)丁-2-烯中间体和共沸溶剂相,下层为过量的烯丙醇相),分相后上层有机相经常压简单蒸馏分出共沸剂后再进行减压蒸馏得到1,1-双(烯丙氧基)丁-2-烯中间体粗品,作为步骤2)裂解重排反应原料。为本领域常规操作,不需要特别限定。
[0027]
本发明中,步骤2)所述裂解重排反应采用反应精馏方式进行,1,1-双(烯丙氧基)丁-2-烯中间体在塔釜同时发生裂解和重排反应,得到2,6-二庚烯醛,塔顶采出烯丙醇,回
收套用。
[0028]
本发明中,步骤2)所述裂解重排反应以间歇或连续方式进行,优选采用连续方式,反应温度为90-150℃,优选110-130℃;停留时间为1-8h,优选3-5h;
[0029]
优选地,所述裂解重排反应在氮气氛围中进行;
[0030]
优选地,所述裂解重排反应,精馏操作压力为1-50kpaa,优选5-30kpaa;回流比1:5-5:1,优选1:2-2:1。
[0031]
本发明中,步骤2)所述裂解重排反应可以在催化剂条件下反应,也可以在无催化剂条件下反应,出于经济性考虑,优选在催化剂条件下反应,所述催化剂为酸催化剂,优选磺酸类化合物,包括但不限于甲基磺酸、乙基磺酸、苯磺酸、对甲苯磺酸等中的一种或多种;
[0032]
所述催化剂基于1,1-双(烯丙氧基)丁-2-烯中间体质量的质量空速为5-100h-1
;
[0033]
在一些具体示例中,在所述裂解重排反应过程中,还优选添加有助剂,所述助剂为锌、铁、锂、锌、钪中的一种或多种的氧化物或盐,包括但不限于氧化锌、氧化铁、氯化锂、溴化锌、甲基磺酸锌、对甲苯磺酸锌、三氟甲磺酸钪等中的一种或多种,优选为溴化锌和/或三氟甲磺酸钪;
[0034]
所述助剂基于1,1-双(烯丙氧基)丁-2-烯中间体质量的质量空速为5-100h-1
。
[0035]
本发明中,步骤2)所述裂解重排反应可以在溶剂或非溶剂环境中进行,优选在溶剂环境中进行;
[0036]
所述溶剂包括但不限于对苯二甲酸二甲酯、n-甲基吡咯烷酮、二甲基甲酰胺、乙二醇二乙酸酯中的一种或多种,优选对苯二甲酸二甲酯和/或n-甲基吡咯烷酮;
[0037]
所述溶剂用量为1,1-双(烯丙氧基)丁-2-烯中间体质量的1-50wt%。
[0038]
本发明中,步骤3)所述选择性加氢反应,温度为50-100℃,优选60-90℃,时间为2.0-6.0h,优选3.0-5.0h;
[0039]
优选地,所述选择性加氢反应在高压反应釜中以间歇或连续方式进行,控制反应压力为0.5-3.0mpag,优选1.0-2.5mpag;反应压力通过通入的氢气控制;
[0040]
本发明中,步骤3)所述选择性加氢反应,可以在催化剂条件下反应,也可以在无催化剂条件下反应,出于经济性考虑,优选在催化剂条件下反应,所述催化剂为负载钯催化剂;
[0041]
优选地,所述负载钯催化剂的载体选自碳酸钙、硫酸钙、硫酸钡中的一种或多种,优选碳酸钙和/或硫酸钡;
[0042]
优选地,所述负载钯催化剂中钯的负载量为0.1-10.0wt%,优选3.0-7.0wt%;
[0043]
所述催化剂用量为2,6-二庚烯醛质量的0.5-5.0wt%,优选1.0-3.0wt%;
[0044]
在一些具体示例中,在所述选择性加氢反应,还优选添加有助剂,所述助剂为叔胺化合物、叔膦化合物中的一种或多种,优选为叔胺化合物和叔膦化合物的混合物,所述叔胺化合物选自三甲胺、三乙胺、三丙胺、三丁胺等中的一种或多种,所述叔膦化合物选自三苯基膦、三(2-甲基苯基)膦、三丁基膦等中的一种或多种;
[0045]
所述助剂用量为2,6-二庚烯醛质量的0.1-0.5wt%,优选0.2-0.4wt%。
[0046]
本发明中,步骤3)所述选择性加氢反应可以在溶剂或非溶剂环境中进行,优选在溶剂环境中进行;
[0047]
所述溶剂包括但不限于乙醇、异丙醇、丙酮、甲苯、乙酸乙酯和四氢呋喃中的一种
或多种,优选甲苯和/或丙酮;
[0048]
所述溶剂用量与2,6-二庚烯醛的质量比为1:0.8-1.2。
[0049]
与现有技术相比,本发明技术方案有益效果在于:
[0050]
本发明提供一种以巴豆醛、烯丙醇为原料通过缩合、重排和加氢合成正庚醛的新方法,原料巴豆醛、烯丙醇廉价易得,合成路线新颖、收率高,具有较好的成本优势;整个合成过程中只产生少量的废水副产,符合绿色化工和原子经济性原则。
具体实施方式
[0051]
下面通过实施例详述本发明,但本发明并不限于下述的实施例。
[0052]
本发明实施例和对比例中使用的主要原料来源信息如下,其余试剂原料如无特别说明,均为普通市售产品:
[0053]
巴豆醛、3-羟基丁醛、3-甲氧基丁醛:安耐吉化学,99%;
[0054]
甲基乙烯基酮、4-羟基-2-丁酮:麦克林生化;
[0055]
丙酮、磷酸三甲酯:西陇试剂;
[0056]
磷酸三乙酯、磷酸三丙酯:萨恩化学;
[0057]
氧化锌、氧化镁、氧化钡:辽宁海泰,95%;
[0058]
5%钯碳、5%钯氧化铝、氧化铂:康纳催化剂;
[0059]
三甲苯:西陇试剂,ar;
[0060]
二叔丁基过氧化物:安耐吉化学;
[0061]
rhcl3、rh(co)2acac、rh4(co)
12
、rh6(co)
16
:麦克林生化。
[0062]
本发明采用的分析方法如下:
[0063]
气相色谱:仪器型号:agilent 7890b;色谱柱:毛细管柱hp-5(30m
×
0.30mm
×
0.25μm);初始温度40℃,以5℃/min的速率升至80℃;再以15℃/min的速率升至240℃,保持5min。载气高纯氮气,分流比30:1,分流流量45ml/min。载气节省:20ml/min,开始等待时间2min。进样温度250℃,检测器为fid,检测器温度250℃,空气流量350ml/min,氢气流量35ml/min,尾吹气流量30ml/min,进样量0.2μl。
[0064]
高分辨质谱仪器:型号:waters xevo g2 qtof;配成溶液直接进样,与气相色谱类似。
[0065]
实施例1-8:
[0066]
由巴豆醛和烯丙醇通过脱水缩合反应制得1,1-双(烯丙氧基)丁-2-烯中间体
[0067]
实施例1
[0068]
氮气氛围中,室温下依次向反应器中加入共沸溶剂正己烷50g、底物巴豆醛(210.0g,3mol)和烯丙醇(382.8g,6.6mol),再加入6.3g二叔丁基过氧化物、0.063g rhcl3快速搅拌后得到澄清液。在压力101kpa、75℃下脱水缩合反应6h,反应过程中正己烷回流带水,并通过分水器不断将反应生成的水移出,待反应体系再无水移出后,停止搅拌,降温,反应液分为两相,上层为产品和正己烷相,下层为过量的烯丙醇相。上层有机相脱除溶剂得1,1-双(烯丙氧基)丁-2-烯中间体。取样用gc色谱分析,巴豆醛转化率为99.2%,1,1-双(烯丙氧基)丁-2-烯选择性为96.5%。
[0069]
实施例2
[0070]
氮气氛围中,室温下依次向反应器中加入共沸溶剂甲苯25g、底物巴豆醛(210.0g,3mol)和烯丙醇(522.0g,9mol),再加入4.2g二叔丁基过氧化物、0.084g rh(co)2acac快速搅拌后得到澄清液。在压力50kpa、90℃下脱水缩合反应4h,反应过程中甲苯回流带水,并通过分水器不断将反应生成的水移出,待反应体系再无水移出后,停止搅拌,降温,反应液分为两相,上层为产品和甲苯相,下层为过量的烯丙醇相。上层有机相脱除溶剂得1,1-双(烯丙氧基)丁-2-烯中间体。取样用gc色谱分析,巴豆醛转化率为99.4%,1,1-双(烯丙氧基)丁-2-烯选择性为96.3%。
[0071]
实施例3
[0072]
氮气氛围中,室温下依次向反应器中加入共沸溶剂二甲苯80g、底物巴豆醛(210.0g,3mol)和烯丙醇(435.0g,7.5mol),再加入2.1g二叔丁基过氧化物、0.042g rh4(co)
12
快速搅拌后得到澄清液。在压力15kpa、100℃下脱水缩合反应8h,反应过程中二甲苯回流带水,并通过分水器不断将反应生成的水移出,待反应体系再无水移出后,停止搅拌,降温,反应液分为两相,上层为产品和二甲苯相,下层为过量的烯丙醇相。上层有机相脱除溶剂得1,1-双(烯丙氧基)丁-2-烯中间体。取样用gc色谱分析,巴豆醛转化率为99.7%,1,1-双(烯丙氧基)丁-2-烯选择性为96.1%。
[0073]
实施例4
[0074]
氮气氛围中,室温下依次向反应器中加入共沸溶剂环己烷100g、底物巴豆醛(210.0g,3mol)和烯丙醇(870.0g,15mol),再加入8.4g二叔丁基过氧化物、0.105g rh6(co)
16
快速搅拌后得到澄清液。在压力60kpa、60℃下脱水缩合反应12h,反应过程中环己烷回流带水,并通过分水器不断将反应生成的水移出,待反应体系再无水移出后,停止搅拌,降温,反应液分为两相,上层为产品和环己烷相,下层为过量的烯丙醇相。上层有机相脱除溶剂得1,1-双(烯丙氧基)丁-2-烯中间体。取样用gc色谱分析,巴豆醛转化率为99.5%,1,1-双(烯丙氧基)丁-2-烯选择性为95.7%。
[0075]
对比实施例5
[0076]
参照实施例1脱水缩合反应方法,不同之处仅在于:不加入催化剂二叔丁基过氧化物,其它操作条件不变。取样分析,巴豆醛转化率为5.1%,1,1-双(烯丙氧基)丁-2-烯选择性为67.9%。
[0077]
对比实施例6
[0078]
参照实施例1脱水缩合反应方法,不同之处仅在于:催化剂二叔丁基过氧化物替换为等量的过氧叔丁醇,其它操作条件不变。取样分析,巴豆醛转化率为87.4%,1,1-双(烯丙氧基)丁-2-烯选择性为33.4%。
[0079]
对比实施例7
[0080]
参照实施例1脱水缩合反应方法,不同之处仅在于:不加入助剂rhcl3,其它操作条件不变。取样分析,巴豆醛转化率为90.9%,1,1-双(烯丙氧基)丁-2-烯选择性为83.2%。
[0081]
对比实施例8
[0082]
参照实施例1脱水缩合反应方法,不同之处仅在于:助剂rhcl3替换为等量的rucl3,其它操作条件不变。取样分析,巴豆醛转化率为93.1%,1,1-双(烯丙氧基)丁-2-烯选择性为87.1%。
[0083]
实施例9-16:
[0084]
1,1-双(烯丙氧基)丁-2-烯中间体通过裂解重排反应制得2,6-二庚烯醛
[0085]
实施例9
[0086]
氮气氛围中,室温下向精馏反应釜中加入溶剂对苯二甲酸二甲酯300g及1.2g甲基磺酸与1.2g溴化锌,将塔顶压力控制在20kpa、回流比1:1。将600g1,1-双(烯丙氧基)丁-2-烯中并以1.25g/min速率连续进料,催化剂质量空速62.5h-1
,助剂质量空速62.5h-1
。130℃下裂解重排反应,反应停留时间4h。塔釜无采出,所有组分通过塔顶采出。塔顶取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为99.7%,2,6-二庚烯醛选择性为98.1%。
[0087]
实施例10
[0088]
氮气氛围中,室温下向精馏反应釜中加入溶剂n-甲基吡咯烷酮400g及2.4g苯磺酸与4.8g溴化锌,将塔顶压力控制在50kpa、回流比1:2。将600g 1,1-双(烯丙氧基)丁-2-烯中并以2.22g/min速率连续进料,催化剂质量空速55.5h-1
,助剂质量空速27.75h-1
。150℃下裂解重排反应,反应停留时间3h。塔釜无采出,所有组分通过塔顶采出。塔顶取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为98.8%,2,6-二庚烯醛选择性为98.6%。
[0089]
实施例11
[0090]
氮气氛围中,室温下向精馏反应釜中加入溶剂n-甲基吡咯烷酮200g及4.8g甲基磺酸与2.4g三氟甲磺酸钪,将塔顶压力控制在30kpa、回流比2:1。将600g 1,1-双(烯丙氧基)丁-2-烯中并以0.66g/min速率连续进料,催化剂质量空速8.25h-1
,助剂质量空速16.5h-1
。120℃下裂解重排反应,反应停留时间5h。塔釜无采出,所有组分通过塔顶采出。塔顶取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为99.9%,2,6-二庚烯醛选择性为97.6%。
[0091]
实施例12
[0092]
氮气氛围中,室温下向精馏反应釜中加入溶剂对苯二甲酸二甲酯600g及3.6g对甲苯磺酸与1.2g三氟甲磺酸钪,将塔顶压力控制在5kpa、回流比5:1。将600g 1,1-双(烯丙氧基)丁-2-烯中并以1.25g/min速率连续进料,催化剂质量空速20.8h-1
,助剂质量空速62.5h-1
。110℃下裂解重排反应,反应停留时间8h。塔釜无采出,所有组分通过塔顶采出。塔顶取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为99.8%,2,6-二庚烯醛选择性为96.2%。
[0093]
对比实施例13
[0094]
参照实施例9裂解重排方法,不同之处仅在于:不加入催化剂甲基磺酸,其它操作条件不变。取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为12.4%,2,6-二庚烯醛选择性为93.3%。
[0095]
对比实施例14
[0096]
参照实施例9裂解重排方法,不同之处仅在于:催化剂甲基磺酸替换为等量的硫酸,其它操作条件不变。取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为61.2%,2,6-二庚烯醛选择性为73.5%。
[0097]
对比实施例15
[0098]
参照实施例9裂解重排方法,不同之处仅在于:不加入助剂溴化锌,其它操作条件不变。取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为95.6%,2,6-二庚烯醛选择性为87.4%。
[0099]
对比实施例16
[0100]
参照实施例9裂解重排方法,不同之处仅在于:助剂溴化锌替换为等量的溴化钠,
其它操作条件不变。取样分析,1,1-双(烯丙氧基)丁-2-烯转化率为97.1%,2,6-二庚烯醛选择性为91.3%。
[0101]
实施例17-24:
[0102]
2,6-二庚烯醛通过选择性加氢反应制得正庚醛
[0103]
实施例17
[0104]
依次向高压釜内加入原料2,6-二庚烯醛100g、溶剂乙醇100g、三甲胺0.3g,最后加入催化剂1.0g负载量5wt%的pd/caco3,得到灰色悬浊液。高压釜内持续通入氢气,反应过程中保持压力恒定2.0mpag。搅拌下升温至80℃后,保持恒温、恒压选择性加氢反应4h。取样gc分析,2,6-二庚烯醛转化率为99.5%,产品正庚醛选择性为98.9%。
[0105]
实施例18
[0106]
依次向高压釜内加入原料2,6-二庚烯醛100g、溶剂甲苯80g、三甲胺0.2g,最后加入催化剂2.0g负载量7wt%的pd/caco3,得到灰色悬浊液。高压釜内持续通入氢气,反应过程中保持压力恒定2.5mpag。搅拌下升温至60℃后,保持恒温、恒压选择性加氢反应6h。取样gc分析,2,6-二庚烯醛转化率为99.4%,产品正庚醛选择性为98.3%。
[0107]
实施例19
[0108]
依次向高压釜内加入原料2,6-二庚烯醛100g、溶剂丙酮90g、三丁胺0.2g、三苯基膦0.2g,最后加入催化剂3.0g负载量3wt%的pd/caco3,得到灰色悬浊液。高压釜内持续通入氢气,反应过程中保持压力恒定3.0mpag。搅拌下升温至90℃后,保持恒温、恒压选择性加氢反应5h。取样gc分析,2,6-二庚烯醛转化率为99.8%,产品正庚醛选择性为99.1%。
[0109]
实施例20
[0110]
依次向高压釜内加入原料2,6-二庚烯醛100g、溶剂甲苯120g、三苯基膦0.5g,最后加入催化剂5.0g负载量9wt%的pd/baso4,得到灰色悬浊液。高压釜内持续通入氢气,反应过程中保持压力恒定1.0mpag。搅拌下升温至100℃后,保持恒温、恒压选择性加氢反应3h。取样gc分析,2,6-二庚烯醛转化率为99.6%,产品正庚醛选择性为99.4%。
[0111]
对比实施例21
[0112]
参照实施例17选择性加氢方法,不同之处仅在于:不加入催化剂pd/caco3,其它操作条件不变。取样分析,2,6-二庚烯醛转化率为1.3%。
[0113]
对比实施例22
[0114]
参照实施例17选择性加氢方法,不同之处仅在于:催化剂pd/caco3替换为等量的雷尼镍催化剂,其它操作条件不变。取样分析,2,6-二庚烯醛转化率为99.8%,产品正庚醛选择性为15.6%。
[0115]
对比实施例23
[0116]
参照实施例17选择性加氢方法,不同之处仅在于:不加入助剂三甲胺,其它操作条件不变。取样分析,2,6-二庚烯醛转化率为99.7%,产品正庚醛选择性为53.1%。
[0117]
对比实施例24
[0118]
参照实施例17选择性加氢方法,不同之处仅在于:助剂三甲胺替换为等量的三乙醇氨,其它操作条件不变。取样分析,2,6-二庚烯醛转化率为37.1%,产品正庚醛选择性为75.8%。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。