1.本发明属于生化科学与计算机化学相结合,涉及人工智能技术领域,特别涉及基于并行式深层细粒度模型预测药物靶标结合性的方法。
背景技术:
2.药物靶标可结合性分析是现代医学中一个非常重要的课题,是认识现有药物功能,探究药物靶标结合机制,扩展药物使用的潜在空间,实现药物再利用的重要方法。当前,应用传统医学实验方法研究药物靶标结合性体现出成本高、失败率高、回报周期长的缺陷,并且在某些情况下由于伦理道德问题而限制实验的开展。随着交叉学科的发展,计算机技术越来越多的应用到化学、生物学等领域,用以解决传统化学、生物学实验方法难以获得甚至无法获得的分子信息,服务于研究药物靶标结合性研究。基于人工智能的方法能够有效整合大量生物分子信息,同时得到生化分子的嵌入特征表达,并使用深度神经网络进行特征学习和归纳,实现对潜在药物靶标作用对的预测,克服了传统实验的缺点和不足。
3.常用的药物靶标结合性预测模型主要分为基于机器学习(machine learning,ml)的模型和基于深度学习(deep learning,dl)的模型。基于机器学习的模型主要通过如矩阵分解、随机森林或支持向量机等方法,达到预测药物靶标结合性的目的。这些方法在数据集规模较小或药物靶标作用关系较简单时可以取得一定效果,但是无法适应较大规模的数据集,且无法精确归纳生物分子特征。而基于深度学习的模型虽然相较于机器学习的预测性能取得了一定的提升,但是简单神经网络无法准确的归纳多种异构信息,同时复杂的神经网络架构如图注意力网络则具有较大的计算开销。此外,先前的方法更多考虑的是药物靶标之间的简单结合关系,并没有将复杂的异构网络信息充分纳入考虑。实际情况中,药物靶标关系间不止存在“一种基因-一种药物-一种疾病”的单一作用模式,因此先前的机器学习和深度学习模型很难取得优异的预测结果。相较于药物靶标单一作用模式,生物分子的异构网络代表更广泛的相互作用关系,其主要体现为相互作用关系复杂、生物分子种类多、数据量庞大等特点。目前较为流行的针对异构网络信息的计算模型,如dtinet(drug-target interactions prediction net)、gcn-dti(graph convolution network)等模型存在对于异构网络信息整合不充分、计算成本高的问题。
技术实现要素:
4.为解决上述现有技术中所存在的针对异构网络信息处理的模型存在计算成本高,异构信息利用不充分问题,本发明提供一种基于并行式深层细粒度模型预测药物靶标结合性的方法,用以快速,高效,准确且低成本的预测药物靶标的结合关系,从而为药物再利用创造条件。
5.为了实现上述技术目的,本发明提供了如下技术方案:
6.基于并行式深层细粒度模型预测药物靶标结合性的方法,包括:
7.获取药物、蛋白质、疾病的数据集,基于数据集,通过并行计算训练预测模型,并对
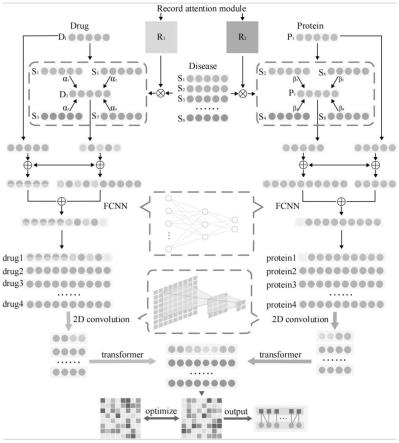
训练好的预测模型进行验证;获取药物靶标数据,通过验证后的预测模型对药物靶标数据进行预测,得到药物靶标结合性预测结果。其中所述预测模型为深层细粒度模型,包括构造器、编码器、解码器;通过构造器提取数据集的本体特征;通过编码器对本体特征进行细粒度级别融合,得到综合性特征,通过解码器对综合性特征进行解码,得到分类预测结果,其中分类预测结果为药物是否存在相互作用。
8.可选的,获取药物、蛋白质、疾病的数据集的具体过程包括:
9.获取药物、蛋白质及疾病的异构网络及同构网络,基于异构网络及同构网络,获取药物、蛋白质、疾病的数据集;其中,所述异构网络包括药物副作用网络、药物和疾病关联网络、蛋白质和疾病关联网络;所述同构网络包括药物和药物相互作用网络、蛋白质和蛋白质相互作用网络、疾病和疾病相关作用网络;
10.可选的,提取数据集的本体特征的具体过程包括:
11.通过基于本体的生物分析特征构建对数据集进行计算得到分子内在特征,对于同构网络,通过重启式随机游走算法在同构网络上进行游走,得到同构网络拓扑结构,对于异构网络,通过杰卡德相似度系数对异构网络进行计算,得到相似度网络,通过重启式随机游走算法在相似度网络上进行游走,得到异构网络拓扑信息,通过对异构特征向量;
12.基于分子内在特征、同构网络拓扑结构及异构网络拓扑信息得到分子本体特征,分子本体特征包括疾病特征、药物特征及蛋白特征。
13.可选的,编码器采用带有记忆单元的图注意力网络。
14.可选的,程包括:分别将疾病特征在细粒度级别融入药物特征及蛋白质特征,融入药物特征及蛋白质特征的过程采用并行计算。
15.可选的,将疾病特征在细粒度级别融入药物特征的过程包括:
16.向带有记忆单元的图注意力网络中载入疾病特征和药物特征,通过记忆模块提取疾病特征的异构信息,通过注意力网络中的系数对药物特征进行更新,将药物特征在细粒度级融合疾病特征的异构信息,得到药物综合性信息。
17.可选的,将疾病特征在细粒度级别融入蛋白特征的过程包括:
18.向带有记忆单元的图注意力网络中载入疾病特征和蛋白特征,通过记忆模块提取疾病特征的异构信息,通过注意力网络中的系数对蛋白特征进行更新,将更新后的蛋白特征在细粒度级融合疾病特征的异构信息,得到蛋白综合性信息。
19.可选的,所述解码器由二维卷积神经网络和transformer耦合而成。
20.可选的,通过解码器对综合性特征进行解码的过程包括:
21.通过对综合性信息中的蛋白综合性信息及药物综合性信息进行特征组合,形成蛋白质和药物作用关系对,通过解码器对蛋白质和药物作用关系对进行解码,得到分类预测结果。
22.本发明具有如下技术效果:
23.(1)本发明通过端到端的深度学习模型计算得到可靠的潜在的药物靶标结合作用情况。
24.(2)pdfdti为如何在大规模数据集上执行时以平衡的方式整合全局信息和局部信息提供解决方案。
25.(3)并行计算机制大大降低了模型的计算复杂性。带有记忆单元的图式注意力网
络为实现细粒度的特征整合提供了一个解决方案。
26.(4)该模型的解码器实现了多个神经网络的协调运行,为网络在dti预测中的耦合应用提供了思路,时间少、成本低且预测精度高,方法成本低廉简便,能够节省大量的人力、物力和财力,为药物靶标预测精确度提供相应的基础工具与快捷途径。
附图说明
27.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
28.图1为进行生物分子本体特征提取的流程图;
29.图2为基于图拓扑网络信息的多重算法聚合构建特征图;
30.图3为细粒度级并行编码解码预测药物靶标结合性流程图;
31.图4为1:1稠密数据集baseline模型及pdfdti性能评估图;
32.图5为1:10半稠密数据集baseline模型及pdfdti性能评估图;
33.图6为1:100稀疏数据集baseline模型及pdfdti性能评估图;
34.图7为去除单一匹配的baseline模型及pdfdti鲁棒性测试图;
35.图8为去除二值匹配的baseline模型及pdfdti鲁棒性测试图;
36.图9为加入数据噪声的baseline模型及pdfdti鲁棒性测试图;
37.图10为增强型稀疏数据集的baseline模型及pdfdti鲁棒性测试图;
38.图11为药物疾病异构融合记忆单元维度与模型表现性能对照曲线图;
39.图12为蛋白质疾病异构融合记忆单元维度与模型表现性能对照曲线图;
40.图13为预测出三种药物潜在药物靶标关系图。
具体实施方式
41.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
42.针对现有技术中,传统医学方法常通过复杂漫长的生化实验研究药物靶标的结合性,该类方法具有实验成本高、失败率较高且回报周期长的缺陷。而通过机器学习的方法无法应对当前大规模的数据集和复杂异构信息的处理。现有的针对异构网络信息处理的模型存在计算成本高,异构信息利用不充分的问题。
43.为实现对异构网络信息充分整合的同时显著降低药物靶标预测成本,本发明提出基于并行式深层细粒度模型用于药物靶标结合性预测。模型携带记忆单元的图注意力网络实现特征提取,将异构信息在生物分子特征提取过程中编码成更可靠的嵌入特征,从而提高药物靶标结合性预测的精确度。同时为解决神经网络学习分子特征非常耗时,计算成本高等问题,本发明采用端到端并行式的神经网络模型。通过基于本体相似度计算方法和基于图拓扑信息的相似度计算方法得到分子特征,后使用解码器-编码器融合异构信息并得
到预测结果,本发明实现了快速且精确地预测药物靶标对,极大的降低了计算分子特征的成本,解决了传统医学方法成本高且失败率高的问题,加快了药物再利用研发进程。
44.本发明的原理是通过并行式架构的耦合神经网络,实现提取生物分子细粒度特征,从而建立一个可用于预测药物靶标结合性的深度学习模型。模型主要由生物分子特征构造器,异构信息并行编码器和异构信息解码器构成,采用的技术方案如下:
45.首先从药物、蛋白质、疾病数据库中选取已有的异构网络和同构网络构建数据集。然后通过并行计算训练pdfdti模型,pdfdti模型中的生物分子特征构造器通过两种方法计算训练集中的生物分子内在相似度,从而提取其嵌入式特征。pdfdti模型中的异构信息并行编码器将疾病分子异构信息在细粒度级别融入药物和蛋白质的特征中。同时pdfdti模型中的异构信息解码器整合局部信息与全局信息,完成药物靶标结合性预测模型训练。最后将数据集中剩余的药物靶标对作为测试集验证pdfdti模型的预测结果,同时对模型进行时间复杂度和鲁棒性的验证。
46.所述异构网络包括:药物副作用网络、药物和疾病关联网络、蛋白质和疾病关联网络。
47.所述同构网络包括:药物和药物相互作用网络、蛋白质和蛋白质相互作用网络、疾病和疾病相互作用网络。
48.所述pdfdti模型中的生物分子特征构造器包括基于生物分子本体的特征构建模块和基于网络拓扑特性的特征构建模块。
49.通过两种方法计算训练集中的生物分子内在相似度包括以下步骤:
50.对于药物,靶标和疾病分子进行基于生物分子本体的特征构建。首先使用基于本体的特征构建获得以化学结构的相似性为基础的生化分子可计算特征,使用重启式的随机游走在同构网络上运行以提取同构作用网络的拓扑信息。其次,通过jaccard相似度系数计算得到异构网络的相似度网络,再对其应用重启式的随机游走算法。最后,将这两种通过不同维度生成的特征向量进行相加并通过前馈神经网络进行降维,得到稠密分布的特征向量。
51.所述对于药物,靶标和疾病分子进行基于生物分子本体的特征构建包括以下步骤:通过tanimoto系数比较不同药物间化学结构的差异性实现,其中原子的差异性也被纳入了比较的范围。通过基于蛋白质主序列的smith-waterman评分来计算蛋白质分子的特征。通过比较疾病dag相似性来计算疾病分子的特征。对于每种疾病,根据层次描述符构建一个有向无环图(dag),其中节点代表疾病mesh描述符(或疾病术语),边代表当前节点和其祖先之间的关系。疾病相似性的计算是通过比较其dag相似性。
52.所述pdfdti模型中的异构信息并行编码器将疾病分子异构信息在细粒度级别融入药物和蛋白质的特征包括以下步骤:
53.异构信息并行编码器接收药物、蛋白质和疾病特征向量作为输入,采用并行计算的方式,同时生成两组携带记忆单元的图注意力神经网络实现同构信息聚合以及异构信息的融合。
54.所述同时生成两组携带记忆单元的图注意力神经网络实现同构信息聚合以及异构信息的融合包括以下步骤:
55.向带有记忆单元的图注意力网络中载入疾病特征和药物特征,通过记忆模块提取
异构信息,再通过注意力网络中的系数对药物向量进行更新,使药物向量细粒度级融合疾病的异构网络信息。实现同一种疾病对于不同的药物特征生成过程提供不同程度的影响。
56.向带有记忆单元的图注意力网络中载入疾病特征和蛋白质特征,设置针对蛋白质特征的记忆模块,进行异构信息提取,再通过注意力网络中的系数对蛋白质向量进行更新,使蛋白质向量细粒度级融合疾病的异构网络信息。实现同种疾病对于不同的蛋白质特征生成过程提供不同程度的影响。
57.所述pdfdti模型中的异构信息解码器整合局部信息与全局信息并完成药物靶标结合性预测包括以下步骤:
58.将不同蛋白质和药物进行特征组合,形成不同的蛋白质和药物作用关系对,输入解码器进行预测。解码器由二维卷积神经网络和transformer耦合而成,用以实现局部信息和全局信息的综合。
59.解码器对不同的药物-蛋白质向量对进行解码,将丰富的特征表征转换为最后的分类预测结果,实现对药物靶标作用关系的预测。
60.所述将数据集中剩余的药物靶标对作为测试集验证pdfdti模型的预测结果包括以下步骤:
61.从数据集中选择剩余的药物靶标对,通过最终模型通过运行训练过程中获得的参数进行预测出的结构等验证pdfdti模型的预测结果。
62.从数据集中选择剩余的药物靶标对,通过使用相同训练集训练的dtinet、bps2vec、dcfme、blmnii、netlaprls、neonet四个深度学习模型在测试集上的表现,与pdfdti模型进行对比验证。
63.所述对模型进行时间复杂度的验证包括以下步骤:
64.使用相同的数据集在dtinet、bps2vec、dcfme、blmnii、netlaprls、neonet等baseline模型上进行预测并记录训练和预测时间,并于pdfdti进行比较,检验pdfdti在减少计算复杂度方面的显著程度。分析对比不同的时间消耗,验证pdfdti模型的计算效率。
65.所述对模型进行鲁棒性验证的包括以下步骤:
66.数据集中冗余的dti被删除,同时增加了一些噪音数据,如未确认的dtis数据。10倍交叉验证法被用来评估基线模型和pdfdti在削减的数据集上的表现。采用了四种测试稳健性的策略来对模型进行综合评价。第一种去除具有相似药物相互作用的冗余dtis(即jaccard相似度》70%)。第二种去除具有相似药物的冗余dtis(即。药物化学结构相似性》70%)或去除与类似药物(即药物化学结构相似性》70%)或同源蛋白质(即蛋白质序列相似性》50%)的冗余dtis。第三种去除与类似疾病的药物或蛋白质的冗余dtis。第四种去除与类似副作用的药物的冗余dtis。
67.本发明的内容在于提供了一种基于异构网络信息整合的细粒度级药物靶标结合性预测模型pdfdti。其中,包括对药物、蛋白质和靶标的特征提取和特征构建,基于并行计算的异构信息融合和结合性预测。
68.为了更好的对上述内容进行说明,本发明通过以下步骤进行具体说明:
69.首先从药物、蛋白质、疾病数据库中选取已有的异构网络和同构网络,包括同构网络和异构网络。同构网络包括药物-药物相互作用网络,从drugbank收集得到,蛋白质-蛋白质相互作用网络,从hprd收集得到。异构网络包括药物副作用异构网络,从sider数据库中
获得,药物蛋白质作用网络从drugbank数据库中获得,疾病蛋白质异构作用网络数据和药物疾病关系数据从ctd数据库中获得。药物、蛋白质和疾病的本体数据分别在获取异构网络和同构网络的过程中同时收集到。
70.收集到的数据进行本体对齐和链接,经过进一步的数据样本清洗后,共计得到708种药物,1512种蛋白质和5603种疾病,包括其本体的化学结构,原子组成及相关化学性质。接下来所获得到的生物分子通过两种方法计算内在的相似度,以提取其嵌入式特征。基于本体的特征提取,药物、靶标和蛋白质分别采取三种不同的方法进行计算,并且经过数据对其的处理流程。基于网络的特征提取重点关注网络拓扑结构对生成特征的影响,对同构网络和异构网络采取不同的网络特征提取算法,最后将两种方法提取到的特征进行融合与降维。处理后的特征进入分类器中实现异构信息的融合和结合性的预测。编码器实现将疾病分子异构信息在细粒度级别融入药物和蛋白质的特征表征中,编码器本身的并行结构加速了模型的处理速度。之后通过模型由多重神经网络耦合得到的解码器进行局部信息与全局信息的整合,完成药物与靶标的结合性分析。模型采用带有权重的交叉熵算法进行参数学习和训练过程。模型与dtinet、bps2vec、dcfme、blmnii、netlaprls、neonet等多个机器学习和深度学习模型的预测效果进行对比,以评估pdfdti模型的应用效果。
71.上述方法的具体步骤为:
72.(1)基于本体的生物分子特征构建过程,如图1所示,
73.第一步药物分子本体特征构建
74.我们从drugbank数据库中提取到708种药物信息,包括其药物化学表达式,化学结构信息,原子化学信息,基本化学性质即药物信息作为本体特征构建所需信息。基于本体的药物特征构建目的是得到药物的嵌入式特征用于神经网络训练,通过计算不同药物同种性质的相似度进行编码。药物本体化学相似度计算通过tanimoto相似度算法进行计算,经过与药物的各种化学结构和的表现方式比较后,选择product-graphs的药物化学结构信息承载对象作为相似度计算的依据,具体的计算公式如下:
[0075][0076]
其中,md为药物本体特征,d为药物特征向量,m表示药物特征向量的维度,n表示药物的数量。
[0077]
第二步蛋白质分子本体特征构建
[0078]
我们从dgidb数据库中提取到1512种蛋白质,包括其蛋白质化学表达式,化学结构信息,原子化学信息,氨基酸序列作为本体特征构建所需材料。与药物本体特征构建类似,基于本体的蛋白质特征构建目的是得到蛋白质的嵌入式特征用于神经网络训练,通过计算不同蛋白质的序列和蛋白质差异性进行编码。蛋白质本体相似度计算通过smith-waterman相似度算法进行计算,通过比较蛋白质的主要氨基酸序列完成。在不同蛋白质中存在相似的氨基酸结构,这些结构无法对蛋白质进行区别,因此相同的结构序列不会被拿入特征构建过程,具体的计算公式如下:
[0079][0080]
其中,m
p
为蛋白本体特征,p为蛋白特征向量,u表示蛋白质特征向量的维度,v表示蛋白质的数量。
[0081]
第三步疾病分子本体特征构建
[0082]
我们从ctd数据库中收集到5603种疾病信息。对于每一种疾病,dag用于区分其与其他疾病的差异性,dag(directed acyclic graph)建立在疾病的分层语义表达基础上,在da图中每一个节点代表药物的mesh描述,边代表当前节点和其祖先节点的联系,通过比较dag结构的相似性构建疾病的本体特征。
[0083][0084]
其中,ms为疾病本体特征,s为疾病特征向量,x表示疾病特征向量的维度,t表示疾病的数量。
[0085]
(2)基于网络拓扑结构的特征构建,如图2所示
[0086]
第一步网络集合分类
[0087]
实验中收集到的网络包括几种网络,分别为药物-药物相互作用网络、蛋白质-蛋白质相互作用网络、疾病-疾病相互作用网络、药物-副作用、药物-疾病和蛋白质-疾病关联网络,根据其网络节点是否为同一种类型的分子,如同为药物分子或疾病分子,将其分为两类,分别是同构网络和异构网络,如下表示:
[0088]hα
={h
drug
,h
protein
,h
disease
}
[0089]hβ
={h
drug-side
,h
drug-disease
,h
protein-disease
}
[0090]
第二步同构网络拓扑信息计算
[0091]
重启式随机游走算法是分析图网络结构常用且准确的算法,我们使用重启式的随机游走算法提取同构网络中蕴藏的拓扑网络信息,其公式表达如下:
[0092][0093]
其中ei表示n维标准基向量,ei(i)=1,ei(j)=0,forall j≠i,pr代表预先定义的重启概率。b代表从节点i到节点的转换概率,公式如下:
[0094][0095]
其中a表示有n个分子的网络的加权相邻矩阵。重启式随机游走算法(random walk with restart,rwr)在每个网络上的表现分别获得了药物、蛋白质和疾病的扩散状态特征。rwr直接在同构网络上运行以提取拓扑信息。
[0096]
针对药物-药物相互作用同构网络,在药物-药物相互作用网络拓扑信息的节点中
载入基于本体构建得到的药物特征,得到更新后的药物特征为:
[0097][0098]
针对蛋白质-蛋白质相互作用同构网络,在蛋白质-蛋白质相互作用网络拓扑信息的节点载入基于本体构建得到的蛋白质特征,得到更新后的蛋白质特征为:
[0099][0100]
针对疾病-疾病相互作用同构网络,在疾病-疾病相互作用网络拓扑信息的节点载入基于本体构建得到的疾病特征,得到更新后的疾病特征为:
[0101][0102]
第三步异构网络拓扑信息构建
[0103]
异构网络上存在性质域完全不同的生物分子节点,因此重启式随机游走算法并不能直接对异构网络进行特征提取,在对异质网络应用rwr之前,首先通过jaccard相似度系数生成相应的相似度网络,实现异构网络的同构化。针对药物-疾病异构网络,计算杰卡德相似度的方法如下:
[0104][0105]
其中,nc表示同时与两种药物相互关联的疾病数量,na和nb分别表示只与第一种或第二种药物相互关联的疾病。
[0106]
对实现同构化的药物-疾病网络使用随机游走算法,根据异构网络拓扑信息,将相关特征载入该节点,生成药物-疾病特征即药物疾病异构信息,并针对药物-副作用异构网络,计算杰卡德相似度的方法如下:
[0107][0108]
其中,表示同时与两种药物相互关联的副作用数量,和分别表示只与第一种或第二种药物相互关联的副作用。
[0109]
对实现同构化的药物-副作用网络使用随机游走算法,生成药物-副作用特征,并针对蛋白质-疾病异构网络,生成蛋白质-疾病特征,计算杰卡德相似度的方法如下:
[0110]
[0111]
其中,表示同时与两种蛋白质相互关联的疾病数量,和分别表示只与第一种或第二种蛋白质相互关联的疾病。
[0112]
对实现同构化的蛋白质-疾病网络使用随机游走算法,根据异构网络拓扑信息,生成蛋白质-疾病特征即蛋白疾病异构信息,并针对蛋白质-疾病异构网络,计算杰卡德相似度的方法如下:
[0113][0114]
其中,表示同时与两种蛋白质相互关联的疾病数量,和分别表示只与第一种或第二种蛋白质相互关联的疾病。
[0115]
(3)细粒度级异构信息融合,如图3所示为细粒级编码解码预测流程。
[0116]
第一步并行计算编码器设置
[0117]
并行计算的编码器由并行计算部分和带有记忆单元的图神经网络构成。并行计算部分预先载入疾病分子异构信息,用于对药物特征和靶标特征进行更新,载入的疾病特征矩阵为90维,根据药物疾病异构信息及蛋白疾病异构信息进行统计,经过数据筛选和清洗,共有90种可以和708种药物和1512种蛋白质发生作用的疾病特征,完成对并行计算部分的设置。
[0118]
所使用的药物特征矩阵为400维,其中共有708种药物特征参与细粒度级疾病异构信息融合与更新,更新前的特征维度为100。所使用的蛋白质矩阵为100维,其中共有1512种蛋白质参与细粒度级疾病异构信息融合与更新。根据药物和蛋白质特征矩阵的维度,分别设置针对药物图注意力网络中的记忆模块,针对蛋白质的图注意力网络中的记忆模块。
[0119]
针对蛋白质的图注意力网络中的记忆模块如下:
[0120][0121]
其中,r
protein
为记忆模块中蛋白特征矩阵,通过本体构建中的更新后的蛋白特征进行构建,r
uv
蛋白质特征向量,u表示蛋白质特征向量的维度,v表示蛋白质的数量。
[0122]
针对药物图注意力网络中的记忆模块如下:
[0123][0124]
其中,r
drug
为记忆模块中蛋白特征矩阵,通过本体构建中的更新后的蛋白特征进行构建,r
nm
蛋白质特征向量,m表示药物特征向量的维度,n表示药物的数量。
[0125]
第二步针对药物的异构信息融合处理
[0126]
对于单个药物而言,其将药物本体特征中的针对单个药物的特征向量可以表示为d=(d1,d2,
…
,dk),其中d表示药物特征的维度,首先根据药物-疾病特征矩阵中筛选出与该药物已被证实的可以发生作用的疾病,并生成mask疾病矩阵s
mask
,其中s
α
,s
β
,
…
,s
γ
表示可以与其相互作用的疾病数量的疾病,在本实验中,疾病特征维度是90维。
[0127]smask
=(s
α
,s
β
,
…
,s
γ
)
[0128]
对于该种药物,使用第一步预先生成的记忆单元与mask疾病矩阵使用哈达玛乘积运算,记忆单元使用凯明分布进行初始化,该记忆单元的值将会在后续过程中进行训练,使其能够携带mask疾病的全局信息,得到具有疾病全局信息的疾病异构记忆单元m
r1
,具体过程如下:
[0129][0130]
接下来,使用该记忆单元与mask疾病特征矩阵再次使用哈达玛乘积运算,使用带有mask疾病的全局信息的记忆单元对mask疾病特征进行更新,使每一种mask疾病都能够蕴含和该种药物能够发生反应的潜在结合模式。
[0131][0132]
由于疾病特征维度和药物特征维度并不统一,为了进行后续计算,使用全连接神经网络对药物和疾病特征矩阵的维度进行统一化,如下所示:
[0133][0134]
经过统一后的疾病特征矩阵和药物特征矩阵的维度都为64维,为了实现在避免传统注意力矩阵高昂计算成本的同时实现细粒度级别的异构信息融合,将药物的本体特征向量在第一个维度上进行复制,得到fd,d为药物特征向量,n为数量,k为药物特征维度,其中第一维的维度与mask疾病特征矩阵中疾病的数量相同。
[0135][0136]
在第二维度上对药物特征矩阵和疾病特征矩阵进行拼接,得到计算后的药物疾病特征对,如下式所示:
[0137]fd1
=sum(concat(m
f0
,m
′
r1
))
[0138]
对单个药物的特征矩阵进行降维,使其第一个维度恢复为与药物特征向量相同的大小,通过在第二维度上进行特征相加,使其第二维度退化为一维,得到细粒度融合后的药物特征向量。
[0139]
fd′
=(d
′1,d
′2,
…
,d
′k)
[0140]
再通过全连接神经网络,使其完全恢复为与药物特征向量相同的维度。
[0141][0142]
考虑到疾病特征向量和药物特征向量处于不同的数值空间,融合后的异构信息融合因子进行维度归一化,以便于后续训练,融合公式如下:
[0143][0144]
通过归一化之后,将异构信息融合因子的数值维持在(0,1)的范围内,并使其与原药物特征向量进行哈达玛乘积运算:
[0145][0146]
为了解决长距离处理过程中,异构信息融合可能导致的本身药物特征信息丰富度的衰减,对融合异构网络信息更新后的药物特征向量和原始药物特征向量进行拼接,得到维度为之前二倍的药物特征向量,即药物-疾病编码后的特征f
dout
,通过全连接神经网络,对其在此进行降维,使其恢复到原始的药物特征向量的维度:
[0147]fdout
=fcnn(concat(f
d3
,fd))
[0148]
第二步针对蛋白质的异构信息融合处理
[0149]
对于单个蛋白质而言,其特征向量可以表示为p=(p
1p
,p2,
…
,pk),其中k表示蛋白质特征的维度,首先根据蛋白-疾病特征从疾病特征矩阵中筛选出与该蛋白质已被证实的可以发生作用的疾病,并生成mask疾病矩阵s
′
mask
,其中s
′
α
,s
′
β
,
…
,s
′
γ
表示可以与其相互作用的疾病数量,在本实验中,疾病特征为64维。
[0150]
s'
mask
=(s
′
α
,s
′
β
,
…
,s
′
γ
)
[0151]
对于该种蛋白质,使用第一步预先生成的记忆单元与mask疾病矩阵使用哈达玛乘积运算,记忆单元使用凯明分布进行初始化,该记忆单元的值将会在后续过程中进行训练,使其能够携带mask疾病的全局信息,得到具有疾病全局信息的疾病异构记忆单元m
r2
,具体过程如下:
[0152][0153]
接下来,使用该记忆单元与mask疾病特征矩阵再次使用哈达玛乘积运算,使用带有mask疾病的全局信息的记忆单元对mask疾病特征进行更新,使每一种mask疾病都能够蕴含和该种蛋白质能够发生反应的潜在结合模式。
[0154][0155]
由于疾病特征维度和蛋白质特征维度并不统一,为了进行后续计算,使用全连接神经网络对蛋白质和疾病特征矩阵的维度进行统一化,如下所示:
[0156][0157]
经过统一后的疾病特征矩阵和蛋白质特征矩阵的维度都为64维,为了实现在避免传统注意力矩阵高昂计算成本的同时实现细粒度级别的异构信息融合,将蛋白质的特征向量在第一个维度上进行复制,得到f
p
,其中第一维的维度与mask疾病特征矩阵中疾病的数量相同。
[0158][0159]
在第二维度上对蛋白质特征矩阵和疾病特征矩阵进行拼接,得到广播药物疾病特征对,如下式所示:
[0160]fp1
=sum(concat(m
f0
,m
′
r2
))
[0161]
对单个蛋白质的特征矩阵进行降维,使其第一个维度恢复为与蛋白质特征向量相同的大小,通过在第二维度上进行特征相加,使其第二维度退化为1,得到细粒度融合后的
蛋白质特征向量。
[0162]fp
′
=(p
′1,p
′2,
…
,p
′k)
[0163]
再通过全连接神经网络,使其完全恢复为与蛋白质特征向量相同的维度,公式如下:
[0164][0165]
考虑到疾病特征向量和蛋白质特征向量处于不同的数值空间,融合后的异构信息融合因子进行维度归一化,以便于后续训练,融合公式如下:
[0166][0167]
通过归一化之后,将异构信息融合因子的数值维持在(0,1)的范围内,并使其与原蛋白质特征向量进行哈达玛乘积运算:
[0168][0169]
为了解决长距离处理过程中,异构信息融合可能导致的本身蛋白质特征信息丰富度的衰减,对融合异构网络信息更新后的蛋白质特征向量和原始药物特征向量进行拼接,得到维度为之前二倍的蛋白质特征向量,即蛋白-药物编码后特征f
pout
,通过全连接神经网络,对其在此进行降维,使其恢复到原始的蛋白质特征向量的维度。
[0170]fpout
=fcnn(concat(f
p3
,f
p
))
[0171]
第三步构造药物-靶标特征对
[0172]
通过拼接运算,将都经过异构信息融合的药物和靶标特征向量经行依此拼接,得到药物和靶标特征对,用于后续解码器的运算:
[0173]
p=concat(f
dout
,f
pout
)
[0174]
(4)预测药物靶标结合性
[0175]
第一步编码后的特征向量的接收和载入
[0176]
模型分类器的解码器获得由编码器组装的药物-蛋白对的综合性特征。解码器接收编码器编码后的药物-靶标特征向量,通过初始化神经网络参数载入药物-靶标特征向量对。
[0177]
第二步二维卷积神经网络整合全局信息
[0178]
首先使用二维卷积神经网络对与药物-靶标特征矩阵进行卷积操作,将卷积核的感受域设置为单个的药物-靶标特征向量对,以对药物-靶标特征向量对的全局信息进行提取:
[0179]
p
′
=2dconvolution(p)
[0180]
第三步使用自定义transformer整合信息并输出结果
[0181]
经过卷积神经网络后的药物-靶标作用对的维度发生变化。通过引用部分transformer的部件进行深度特征融合,并输出最后dti预测结果:
[0182]sd,t
=transforme r(p
′
)
[0183]
dti预测为二分类任务,设置是否存在相互作用的阈值为0.5,当dti预测出的值大于0.5时,将其归纳到预测为存在相互作用关系的药物靶标对,反之归纳为不存在相互作用关系的药物-靶标对:
[0184][0185]
如图13所示为预测的三种药物潜在药物靶标关系。
[0186]
(5)模型训练与使用
[0187]
第一步使用带有权重的交叉熵算法优化模型参数
[0188]
数据集中的药物-目标对被定义为阳性样本和阴性样本。n和m分别表示给定数据集中的药物和蛋白质的数量。
[0189]
在此,正面实例的集合和负面实例的集合分别表示为y
和y-。dtis预测被认为是一个二元任务。
[0190]
然而,关联的数量比没有观察到关联的药物-目标对要少得多。在此,我们采用加权交叉熵损失函数,即pddat,具体如下
[0191][0192]
其中a
ij
分别表示一对药物di和蛋白质pj。式中|y
|和|y-|分别为y
和y-的实例数。权重系数lambda规定了观察到的关联的重要性,以减少数据不平衡的影响。
[0193]
第二步确定预测模型评价指标
[0194]
f1:综合考虑到准确度和召回率的影响。
[0195][0196]
acc:重点关注于模型的预测准确度能力。
[0197][0198]
sen:灵敏度表示所有正例中,被分对的比例,衡量给了分类器对正例的识别能力。
[0199][0200]
spe:特效度表达了对负例的识别能力,和灵敏度相似。
[0201][0202]
pre:精度是精确性的度量,表示被分为正例的示例中,实际为正例的比例。
[0203][0204]
第三步实验数据集的划分和选择
[0205]
我们在三个具有不同比例的阳性和阴性样本的数据集上进行了实验。
[0206]
在这些数据集中,阳性样本是所有已知的药物-目标关联,阴性样本是随机产生的药物目标对。两个数据集的阳性和阴性样本比例分别为1:1、1:10和1:100。
[0207]
采用10倍交叉验证来评估数据集上的每个模型。在每个10倍交叉验证实验中,数据集被随机分割成90%的阳性和阴性子集作为训练集,并将其余10%的样本作为测试集。为了减少交叉验证的数据偏差,实验进行了10次,并计算出结果的平均值。
[0208]
第四步多种模型的训练
[0209]
从github下载baseline模型并根据其他研究报告设置参数细节,分别设置dtinet、bps2vec、dcfme、blmnii、netlaprls、neonet的具体参数和载入与实验模型所用相同的数据集,分别将1:1,1:10,1:100的数据集进行载入。
[0210]
设置pdfdti的模型参数,载入1:1、1:10和1:100的相关数据集。
[0211]
使用基于linux 18.02内核版本的ubuntu操作系统进行训练,所使用显卡为rtx3080显卡,对模型分部实验并记录各个模型的表现情况。
[0212]
根据其训练时表现结果,动态对模型的超参数进行调节,最大程度提高模型的预测性能,保存并记录模型最优的参数值。在对记忆单元维度进行调节后,模型表现性能如图11-12所示。
[0213]
第五步不同模型性能分析
[0214]
如图4-10所示,pdfdti优于其他比较方法,在1:1的比例下,pdfdti分别比亚军方法获得了多出5.9%的f1得分和5.8%的aupr得分;在1:10的比例下,pdfdti比亚军方法获得了多出8.7%的f1得分和5.9%的aupr得分。
[0215]
为了模拟真实环境,我们将正负样本的比例扩大到1:100,其中负样本是正样本的100倍。还进行了10倍的交叉验证。pdfdti在f1值比所有基线方法至少多出6.4%的分数,在aupr方面则多出7.1%的分数。
[0216]
以上显示和描述了本发明的基本原理、主要特征和优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。