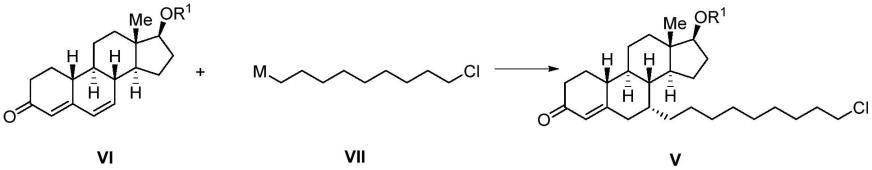
1.本发明涉及一种用于制备氟维司群的中间体及其制备方法。
背景技术:
2.氟维司群(如式i所示)是一种雌激素受体拮抗剂。2002年4月,阿斯利康的氟维司群注射液被fda批准上市,2018年时其全球销售额已经超过了10亿美元。
[0003][0004]
现有技术对氟维司群的合成主要通过对甾体母核的c7位引进侧链的方式来进行,侧链的引入方法大致上分为烷基化和格氏试剂加成两种策略。
[0005]
如专利cn102827048、cn102993257、cn110938107、cn102993259、wo2009013310、wo2009039700、us9315540中所公开,烷基化路线以雌二醇为原料,经保护后,需要先对c-6氧化形成醇,再氧化形成酮。进行烷基化反应后,又需要将酮还原为烷基,以获得氟维司群母体结构,反应操作比较繁琐。
[0006][0007]
格氏加成路线以脱氢诺龙醋酸酯为原料,经过格氏加成和芳构化两步反应得到氟维司群母体结构。在格氏试剂的结构上,专利wo2002032922、wo2006015081公开了当r为
的格氏试剂进行加成的路线。虽然路线十分简洁,但是在其相应的文献organic process research&development 2010,14,544-552中提到,当结构中同时含有硫和溴时,其格氏试剂的前体溴代物不稳定,易于降解,且少量杂质的存在也会导致格氏试剂的引发困难,因此对物料的保存和纯度的要求非常高。
[0008][0009]
wo2006015081和cn103965280b还公开了当r为otbs时,相对而言短链的格氏试剂对脱氢诺龙醋酸酯加成的方案。但是在该情况下,需要将叔丁基二甲基硅烷保护基水解脱除并转化为离去基团后,才能引入五氟戊硫醇片段,从而增加了合成路线的长度和纯化的难度。
[0010][0011]
综上,现有制备氟维司群的方法需要对载体骨架进行反复的氧化还原反应,或者对侧链进行反复的保护和去保护反应,导致反应路线比较繁琐,纯化困难,合成效率低等缺点。因此,需要开发一种更高效的合成路线来制备氟维司群。
技术实现要素:
[0012]
针对现有氟维司群制备方法的缺点,经过发明人的不懈努力,发明了一种制备氟维司群的新中间体和合成方法,以及经由该中间体合成氟维司群的制备方法,该方法合成
路线简短,反应条件温和安全,操作和纯化简便,反应选择性高,合成效率高,适于大规模生产氟维司群。
[0013]
本发明提供一种如式v所示的化合物,
[0014][0015]
其中,r1为氢或羟基保护基。
[0016]
本发明一个优选的实施方案,式v所示的化合物中,r1为氢、c
1-c6烷基、c
1-c
10
烷基酰基、芳基酰基或(c
1-10
烷基或芳基)3硅烷基。
[0017]
本发明一个更优选的实施方案,式v所示的化合物中,r1为c
1-c
10
烷基酰基或芳基酰基。
[0018]
本发明一个更优选的实施方案,式v所示的化合物中,r1为乙酰基。
[0019]
本发明还提供一种如式v所示的化合物的制备方法,其包括如下步骤:将式vi所示的化合物与式vii所示的化合物经过加成反应得到如式v所示的化合物;所述加成反应优选在铜试剂的催化下进行;
[0020][0021]
其中,r1为氢或羟基保护基;m为mgcl、mgbr、mgi、mg
1/2
、zncl、znbr、zni、zn
1/2
、mncl、li或na。
[0022]
本发明一个优选的实施方案,所述的如式v所示化合物的制备方法中,所述的r1可为氢、c
1-c6烷基、c
1-c
10
烷基酰基、芳基酰基或(c
1-10
烷基或芳基)3硅烷基;m为mgcl或mgbr。
[0023]
本发明一个更优选的实施方案,所述的如式v所示化合物的制备方法中,所述的r1可为c
1-c
10
烷基酰基或芳基酰基;m为mgcl或mgbr。
[0024]
本发明一个更优选的实施方案,所述的如式v所示化合物的制备方法中,所述的r1可为乙酰基;m为mgbr。
[0025]
所述的如式v所示化合物的制备方法中,所述的加成反应产生一组非对映异构体混合物。作为示例地,所述非对映异构体v-7α:v-7β的比例可为100~1:1。所述非对映异构体混合物可以直接进行下一步反应,也可以经过纯化后得到单一的异构体再进行下一步反应。
[0026][0027]
所述的如式v所示化合物的制备方法中,所述的加成反应可在有机溶剂中进行。所述的有机溶剂可为本领域该类反应的常规溶剂,还可为c
6-c
10
烷烃溶剂(如正己烷、正庚烷)、芳烃类溶剂(如甲苯)、醚类溶剂(如四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、二噁烷)中的一种或多种。所述的有机溶剂的用量可为本领域该类反应的常规用量,例如所述的式vi所示的化合物在所述的有机溶剂中的摩尔浓度可为0.001~5mol/l。
[0028]
所述的如式v所示化合物的制备方法中,所述的式vi所示的化合物在所述的有机溶剂中的摩尔浓度可进一步优选为0.005、0.01、0.05、0.1、0.5、1.0或5.0mol/l。
[0029]
所述的如式v所示化合物的制备方法中,所述的铜试剂可为氯化亚铜、溴化亚铜、碘化亚铜、三氟甲磺酸亚铜中的一种或多种。所述的铜试剂的用量可为本领域该类反应的常规用量,例如所述的铜试剂与所述的如式vi所示的化合物的摩尔比例可为0.001~1:1。
[0030]
所述的如式v所示化合物的制备方法中,所述的铜试剂与所述的如式vi所示的化合物的摩尔比例可进一步优选为0.01:1、0.05:1、0.1:1、0.3:1、0.5:1或1:1。
[0031]
所述的如式v所示的化合物的制备方法中,所述的式vii所示的化合物与所述的式vi所示的化合物的摩尔比例可为1~10:1。
[0032]
所述的如式v所示的化合物的制备方法中,所述的式vii所示的化合物与所述的式vi所示的化合物的摩尔比例进一步优选为5:1、4:1、3:1、2:1或1:1。
[0033]
所述的如式v所示的化合物的制备方法中,所述的加成反应的进程可以采用本领域中的常规测试方法(如tlc、hplc、gc或nmr,较佳地为hplc)进行监控,一般以所述的如式vi所示的化合物不再反应时作为反应终点。
[0034]
所述的如式v所示的化合物的制备方法中,所述的加成反应的反应温度可为本领域该类反应的常规温度,例如为-50~50℃。
[0035]
所述的如式v所示的化合物的制备方法中,所述的加成反应的反应温度进一步优选为-30℃、-20℃、-10℃、0℃、10℃或20℃。
[0036]
本发明还提供一种如式vii所示的化合物,
[0037][0038]
其中,m为mgcl、mgbr、mgi、mg
1/2
、zncl、znbr、zni、zn
1/2
、mncl、li或na。
[0039]
本发明一个优选的实施方案,m为mgcl或mgbr。
[0040]
本发明一个更优选的实施方案,m为mgbr。
[0041]
本发明还提供一种如式vii所示的化合物的制备方法,其包括如下步骤:将如式viii所述的化合物与金属反应,或者与金属有机化合物发生交换反应制得如式vii所示的化合物,
[0042][0043]
其中,x为氯、溴或碘;m为mgcl、mgbr、mgi、mg
1/2
、zncl、znbr、zni、zn
1/2
、mncl、li或na。
[0044]
本发明一个优选的实施方案,所述的如式vii所示化合物的制备方法中,所述的x为氯或溴,m为mgcl或mgbr。
[0045]
本发明一个更优选的实施方案,所述的如式vii所示化合物的制备方法中,所述的x为溴,m为mgbr。
[0046]
本发明一个优选的实施方案,所述的如式vii所示化合物的制备方法中,如式viii所述的化合物与金属镁发生格式反应制得如式vii所示的化合物,所述的金属镁可为镁粉、镁屑中的一种或多种。
[0047]
所述的如式vii所示化合物的制备方法中,所述的格氏反应可在有机溶剂中进行。所述的有机溶剂可为本领域该类反应的常规溶剂,例如,可为醚类溶剂(如四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、乙醚、二噁烷)中的一种或多种。所述的有机溶剂的用量可为本领域该类反应的常规用量,例如所述的式viii所示的化合物在所述的有机溶剂中的摩尔浓度可为0.001~5mol/l。
[0048]
所述的如式vii所示化合物的制备方法中,所述的式viii所示的化合物在所述的有机溶剂中的摩尔浓度可进一步优选为0.005、0.01、0.05、0.1、0.5、1.0或5.0mol/l。
[0049]
所述的如式vii所示的化合物的制备方法中,所述的金属镁与所述的式viii所示的化合物的摩尔比例可为0.5~10:1。
[0050]
所述的如式vii所示的化合物的制备方法中,所述的金属镁与所述的式viii所示的化合物的摩尔比例进一步优选为0.5:1、0.8:1、0.9:1、0.95:1、1:1、1.5:1、2:1、3:1或5:1。
[0051]
所述的如式vii所示的化合物的制备方法中,所述的格氏反应的反应温度可为本领域该类反应的常规温度,例如为-20~80℃。
[0052]
所述的如式vii所示的化合物的制备方法中,所述的格氏反应的反应温度进一步优选为-10℃、0℃、10℃、20℃、30℃、40℃、50℃或80℃。
[0053]
本发明还提供一种如式iii所示的化合物,
[0054][0055]
其中,r1为氢或羟基保护基;r2为羟基保护基。
[0056]
本发明一个优选的实施方案,式iii所示的化合物中,r1为氢、c
1-c6烷基、c
1-c
10
烷基酰基、芳基酰基或(c
1-10
烷基或芳基)3硅烷基;r2为c
1-c
10
烷基酰基或芳基酰基。
[0057]
本发明一个更优选的实施方案,式iii所示的化合物中,r1为c
1-c
10
烷基酰基或芳
基酰基;r2为c
1-c
10
烷基酰基或芳基酰基。
[0058]
本发明一个更优选的实施方案,式iii所示的化合物中,r1为乙酰基;r2为乙酰基或丙酰基。
[0059]
本发明还提供一种如式iii所示的化合物的制备方法,其包括如下步骤:将如式v所示的化合物通过芳构化反应和酚羟基保护反应制得如式iii所示的化合物;所述芳构化反应优选在铜盐的作用下进行;
[0060]
其中,
[0061]
r1为氢或羟基保护基;r2为羟基保护基。
[0062]
本发明一个优选的实施方案,所述的如式iii所示化合物的制备方法中,所述的r1可为氢、c
1-c6烷基、c
1-c
10
烷基酰基、芳基酰基或(c
1-10
烷基或芳基)3硅烷基;r2为c
1-c
10
烷基酰基或芳基酰基。
[0063]
本发明一个更优选的实施方案,所述的如式iii所示化合物的制备方法中,所述的r1可为选为c
1-c
10
烷基酰基或芳基酰基;r2为c
1-c
10
烷基酰基或芳基酰基。
[0064]
本发明一个更优选的实施方案,所述的如式iii所示化合物的制备方法中,所述的r1可为乙酰基;r2为乙酰基或丙酰基。
[0065]
本发明一个更优选的实施方案,所述的如式iii所示化合物的制备方法中,所述的r1可为乙酰基;r2为乙酰基。
[0066]
所述的如式iii所示化合物的制备方法中,所述的芳构化反应可在有机溶剂中进行。所述的有机溶剂可为本领域该类反应的常规溶剂,还可为腈类溶剂(如正乙腈、丙腈、丁腈、苯腈)、酰胺类溶剂(如n,n-二甲基甲酰胺、n,n-二甲基乙酰胺)、砜类溶剂(如二甲基亚砜、环丁砜)、芳烃类溶剂(如甲苯)、醚类溶剂(如四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、二噁烷)中的一种或多种。所述的有机溶剂的用量可为本领域该类反应的常规用量,例如所述的式v所示的化合物在所述的有机溶剂中的摩尔浓度可为0.001~5mol/l。
[0067]
所述的如式iii所示化合物的制备方法中,所述的式v所示的化合物在所述的有机溶剂中的摩尔浓度可可进一步优选为0.005、0.01、0.05、0.1、0.5、1.0或5.0mol/l。
[0068]
所述的如式iii所示化合物的制备方法中,所述的铜盐可为氯化铜、溴化铜、碘化铜、硫酸铜、醋酸铜、硝酸铜、三氟甲磺酸铜中的一种或多种。所述的铜盐的用量可为本领域该类反应的常规用量,例如所述的铜盐与所述的如式v所示的化合物的摩尔比例可为0.5~10:1。
[0069]
所述的如式iii所示化合物的制备方法中,所述的铜盐与所述的如式v所示的化合物的摩尔比例可进一步优选为0.5:1、0.8:1、0.9:1、1:1、2:1、3:1或5:1。
[0070]
所述的如式iii所示化合物的制备方法中,所述的酚羟基保护反应在酰化试剂作用下完成。所述的酰化试剂可以烷基酰化试剂(如乙酸酐、乙酰氯、丙酸酐、丙酰氯)或芳基
酰化试剂(如苯甲酰氯、苯甲酸酐、对甲基苯甲酰氯、对甲基苯甲酸酐)中的一种或多种。所述的酰化试剂的用量可为本领域该类反应的常规用量,例如所述的酰化试剂与所述的如式v所示的化合物的摩尔比例可为1~10:1。
[0071]
所述的如式iii所示化合物的制备方法中,所述的酰化试剂的用量可为本领域该类反应的常规用量,例如所述的酰化试剂与所述的如式v所示的化合物的摩尔比例可可进一步优选为1:1、2:1、3:1、5:1或8:1。
[0072]
所述的如式iii所示的化合物的制备方法中,所述的芳构化反应的进程可以采用本领域中的常规测试方法(如tlc、hplc、gc或nmr,较佳地为hplc)进行监控,一般以所述的如式v所示的化合物不再反应时作为反应终点。
[0073]
所述的如式iii所示的化合物的制备方法中,所述的芳构化反应的反应温度可为本领域该类反应的常规温度,例如为-20~60℃。
[0074]
所述的如式iii所示的化合物的制备方法中,所述的芳构化反应的反应温度可进一步优选为-10℃、0℃、10℃、20℃、30℃、40℃、50℃或60℃。
[0075]
本发明还提供一种如式i所示的氟维司群的制备方法,具体来说,包含如下步骤,
[0076][0077]
其中,r1为氢或羟基保护基;r2为羟基保护基;m为mgcl、mgbr、mgi、mg
1/2
、zncl、znbr、zni、zn
1/2
、mncl、li或na。
[0078]
1)如式vi所示的化合物和式vii所示的化合物经过加成反应得到如式v所示的化合物;所述加成反应优选在铜试剂的作用下进行;所述铜试剂优选为氯化亚铜、溴化亚铜、碘化亚铜、三氟甲磺酸亚铜中的一种或多种;
[0079]
2)如式v所示的化合物经过芳构化反应和酚羟基保护反应得到如式iii所示的化合物;所述芳构化反应优选在铜盐的作用下进行;所述铜盐优选为氯化铜、溴化铜、碘化铜、硫酸铜、醋酸铜、硝酸铜、三氟甲磺酸铜中的一种或多种;所述的酚羟基保护反应在酰化试剂作用下完成,所述的酰化试剂为乙酸酐或丙酸酐;
[0080]
3)如式iii所示的化合物和式iv所示的化合物经过取代反应得到如式ii所示的化合物;所述取代反应优选在碱的作用下进行;所述碱优选为氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾、碳酸铯中的一种或多种;
[0081]
4)如式ii所示的化合物经过氧化反应得到如式i所示的化合物;所述氧化反应优选在氧化剂的作用下进行;所述氧化剂优选为双氧水、间氯过氧苯甲酸、过氧叔丁醇、过氧乙酸中的一种或多种;
[0082]
本发明一个优选的实施方案,所述的如式i所示化合物的制备方法中,所述的r1可为氢、c
1-c6烷基、c
1-c
10
烷基酰基、芳基酰基或(c
1-10
烷基或芳基)3硅烷基;r2为c
1-c
10
烷基酰基或芳基酰基;m为mgcl或mgbr。
[0083]
本发明一个更优选的实施方案,所述的如式i所示化合物的制备方法中,所述的r1可为c
1-c
10
烷基酰基或芳基酰基;r2为c
1-c
10
烷基酰基或芳基酰基;m为mgcl或mgbr。
[0084]
本发明一个更优选的实施方案,所述的如式i所示化合物的制备方法中,所述的r1可为乙酰基;r2为乙酰基或丙酰基;m为mgbr。
[0085]
本发明的一个更优选实施方案,所述的如式i所示的氟维司群的制备方法中,包含如下步骤,
[0086]
1)如式via所示的化合物和式viia所示的化合物经过格氏加成反应得到如式va所示的化合物;
[0087]
2)如式va所示的化合物经过芳构化反应和通过乙酸酐对酚羟基保护反应得到如式iiia所示的化合物;
[0088]
3)如式iiia所示的化合物和式iv所示的化合物经过取代反应得到如式ii所示的化合物;
[0089]
4)如式ii所示的化合物经过氧化反应得到如式i所示的化合物。
[0090]
进一步优选的,包含如下步骤,
[0091]
1)如式via所示的化合物和式viia所示的化合物在氯化亚铜的作用下经过格氏加成反应得到如式va所示的化合物;
[0092]
2)如式va所示的化合物在溴化铜和溴化锂的作用下经过芳构化反应和通过乙酸酐对酚羟基保护反应得到如式iiia所示的化合物;
[0093]
3)如式iiia所示的化合物和式iv所示的化合物在氢氧化钠的作用下经过取代反应得到如式ii所示的化合物;
[0094]
4)如式ii所示的化合物在双氧水和过氧乙酸的作用下经过氧化反应得到如式i所示的化合物;
[0095][0096]
另一方面,本发明还提供了经过如式一种制备氟维司群的方法,包括经1,9-二氯壬烷、1-氯-9-溴壬烷或1-氯-9-碘壬烷制备氟维司群的步骤,
[0097][0098]
其中,1,9-二氯壬烷或1-氯-9-溴壬烷可购得;
[0099]
1-氯-9-溴壬烷也可以通过9-溴-1-壬醇的氯代制得。
[0100]
1-氯-9-碘壬烷的制备可参考文献:j.org.chem.1963,28,1254-1259。
[0101]
另外,化合物iv可购得,或依据文献wo2008044033制得。
[0102]
化合物vi的合成可参考文献:canadian journal of chemistry 1984,62,2740
–
2747。即采用四氯苯醌氧化诺龙得到脱氢诺龙,然后对羟基进行保护制得;
[0103]
[0104]
也可以对脱氢诺龙醋酸酯脱保护后,再更换其他保护基制得;
[0105][0106]
如果可以购得,也可使用上述反应步骤中的部分产物依更短路线制得式v所示化合物;例如可通过购买前述式vi,而后依照上述方法中提供的步骤制得式v所示的化合物。
[0107]
另一方面,本发明还提供了一种制备氟维司群的方法,该方法先依照本发明前述提供的方法制得式v或式iii所示的化合物,而后依照已知的方法经式v或式iii所示的化合物制得氟维司群,所述方法可参照文献:wo2002032922;wo2006015081;organic process research&development 2010,14,544-552。
[0108]
本发明的有益效果是:
[0109]
1)本发明采用末端含氯的金属试剂vii(特别是末端含氯的格式试剂viia)进行加成反应,所得的产物v经过芳构化之后可以直接以氯为离去基团,引入五氟戊硫醇片段,从而避免了现有技术中对保护基的使用和官能团的转化,缩短了氟维司群合成路线的步骤。
[0110]
2)本发明采用末端含氯的格氏试剂viia进行加成反应时反应选择性高,产生的非对映异构体比例可以达到9:1。
[0111]
3)本发明的条件温和,不存在超低温(如小于-40℃)和超高温(如大于80℃)的反应条件。
[0112]
4)本发明的合成操作过程简单,可以不通过柱层析得到合格产品,易于实现工业化放大生产。
[0113]
5)本发明的氟维司群的合成效率具有显著优势。
[0114]
本发明所使用的术语,除有相反的表述外,具有如下的含义:
[0115]
本发明的羟基保护基是本领域已知的适当的用于羟基保护的基团,参见文献(“protective groups in organic synthesis”,5
th ed.t.w.greene&p.g.m.wuts)中的羟基保护基团。作为示例地,所述的羟基保护基可以是(c
1-10
烷基或芳基)3硅烷基,例如:三乙基硅基,三异丙基硅基,叔丁基二甲基硅基,叔丁基二苯基硅基等;可以是c
1-10
烷基或取代烷基,例如:甲基,叔丁基,烯丙基,苄基,甲氧基甲基,乙氧基乙基,2-四氢吡喃基(thp)等;可以是(c
1-10
烷基或芳香基)酰基,例如:甲酰基,乙酰基,苯甲酰基,对甲基苯甲酰基等;可以是(c
1-6
烷基或c
6-10
芳基)磺酰基;也可以是(c
1-6
烷氧基或c
6-10
芳基氧基)羰基。
[0116]“烷基”指饱和的脂族烃基团,包括1至10个碳原子的直链和支链基团,优选包括1至6个碳原子。非限制性实施例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基等。烷基可以是取代的或未取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,优选为一个或多个以下基团,独立地选自烷基、烯基、炔基、烷基氧基、烷硫基、烷基氨基、卤素、硫醇、羟基、硝基、氰基、环烷基、杂环烷基、芳基、杂芳基、环烷基氧基、杂环烷基氧基、环烷硫基、杂环烷硫基或氧代;非限制性实施例包
括但不限于甲基氧基甲基、乙基氧基甲基、苄基、对甲氧基苄基、对甲基苄基、2-四氢吡喃基等。
[0117]“芳基”指具有共轭的π电子体系的6至14元全碳单环或稠合多环(也就是共享毗邻碳原子对的环)基团,优选为6至10元,更优选苯基和萘基,最优选苯基。芳基可以是取代的或未取代的,当被取代时,取代基优选为一个或多个以下基团,独立地选自烷基、烯基、炔基、烷氧基、烷硫基、烷基氨基、卤素、硫醇、羟基、硝基、氰基、环烷基、杂环烷基、芳基、杂芳基、环烷氧基、杂环烷氧基、环烷硫基或杂环烷硫基。
[0118]
缩写表:
[0119]
缩写全称mtbe甲基叔丁基醚ac乙酰基tbs叔丁基二甲基硅基bz苯甲酰基bn苄基mom甲基氧基甲基thp2-四氢吡喃基tso-对甲基苯磺酸根hplc液相色谱
[0120]
下表为实施例中所涉及的化合物的结构式
[0121]
[0122]
[0123]
具体实施方式
[0124]
以下将结合具体实例详细地解释本发明,使得本专业技术人员更全面地理解本发明,具体实例仅用于说明本发明的技术方案,并不以任何方式限定本发明。
[0125]
实施例1:制备化合物viiia
[0126]
将化合物ixa(50g)和氯化亚砜(50ml)加入500ml三口瓶中。控制内温在20℃以下,滴加入吡啶(10ml),反应继续在20℃反应48h。tlc显示原料反应完全。将反应液冰水中猝灭,mtbe萃取,有机相用饱和碳酸氢钠洗涤,干燥浓缩后得52.6g化合物viiia,收率97%。
[0127]1h nmr(400mhz,chloroform-d)δ3.53(td,j=6.7,0.9hz,2h),3.41(td,j=6.9,0.8hz,2h),1.85(p,j=7.0hz,2h),1.81
–
1.72(m,2h),1.43(dq,j=12.9,6.8hz,4h),1.32(d,j=4.5hz,6h).
[0128]
实施例2:制备化合物viia
[0129]
将镁屑(6.42g)加入反应瓶中,加入无水四氢呋喃(380ml)。20℃下加入化合物viiia(46.1g)。滴加完毕后,反应继续在室温下搅拌2h,得到化合物viia,备用。
[0130]
实施例3:制备化合物viia
[0131]
将化合物viiia(46.1g)加入反应瓶中,加入无水四氢呋喃(380ml)。降温至0℃,滴加异丙基溴化镁四氢呋喃溶液(1.0eq)。滴加完毕后,反应继续搅拌2h,得到化合物viia,备用。
[0132]
实施例4:制备化合物viib
[0133]
将镁屑(6g)加入反应瓶中,加入无水四氢呋喃(380ml)。20℃下加入化合物viiib(48g)。滴加完毕后,反应继续在室温下搅拌2h,得到化合物viib,备用。
[0134]
实施例5:制备化合物viic
[0135]
将镁屑(6g)加入反应瓶中,加入无水四氢呋喃(380ml)。20℃下加入化合物viiic(32.7g)。滴加完毕后,反应继续在室温下搅拌2h,得到化合物viib,备用。
[0136]
实施例6:制备化合物viid
[0137]
将镁屑(6.42g)加入反应瓶中,加入无水四氢呋喃(380ml)。20℃下加入化合物viiia(46.1g)。滴加完毕后,反应继续在室温下搅拌2h,得到化合物viia。将viia溶液降温至0℃,加入无水zncl2四氢呋喃溶液(1.0eq),继续搅拌2h,得到化合物viid,备用。
[0138]
实施例7:制备化合物viie
[0139]
将化合物viiia(46.1g)加入反应瓶中,加入无水四氢呋喃(380ml)。降温至-30℃,滴加二乙基锌四氢呋喃溶液(0.5eq)。滴加完毕后,反应继续搅拌2h,得到化合物viie,备
用。
[0140]
实施例8:制备化合物viif
[0141]
将化合物viiia(46.1g)加入反应瓶中,加入无水四氢呋喃(380ml)。降温至-30℃,滴加特丁基锂四氢呋喃溶液(2.5eq)。滴加完毕后,反应继续搅拌2h,得到化合物viif。-30℃下保存备用。
[0142]
实施例9:制备化合物viig
[0143]
将镁屑(6.42g)加入反应瓶中,加入无水四氢呋喃(380ml)。20℃下加入化合物viiia(46.1g)。滴加完毕后,反应继续在室温下搅拌2h,得到化合物viia。将viia溶液降温至0℃,加入无水二氯化锰四氢呋喃溶液(1.0eq),继续搅拌2h,得到化合物viig,备用。
[0144]
实施例10:制备化合物va
[0145]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-20℃下,滴入化合物viia的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-20℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得28.0g化合物va,收率92%。非对映异构体比例7α:7β=9:1。
[0146]
ms(esi)m/z:477(m h
)。
[0147]1h nmr(400mhz,chloroform-d)δ5.82(s,1h),4.61(t,1h),3.52(t,2h),2.52(dd,j=14hz,j=2hz,1h),2.38~2.42(m,1h),2.21~2.29(m,3h),2.15~2.21(m,1h),2.03(s,3h),1.96~2.02(m,1h),1.85~1.88(m,1h),1.68~1.79(m,5h),1.46~1.60(m,4h),0.95~1.42(m,18h).
[0148]
实施例11:制备化合物va
[0149]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-20℃下,滴入化合物viib的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-20℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得25.2g化合物va,收率83%。非对映异构体比例7α:7β=8:1。
[0150]
ms(esi)m/z:477(m h
)。
[0151]1h nmr(400mhz,chloroform-d)δ5.82(s,1h),4.61(t,1h),3.52(t,2h),2.52(dd,j=14hz,j=2hz,1h),2.38~2.42(m,1h),2.21~2.29(m,3h),2.15~2.21(m,1h),2.03(s,3h),1.96~2.02(m,1h),1.85~1.88(m,1h),1.68~1.79(m,5h),1.46~1.60(m,4h),0.95~1.42(m,18h).
[0152]
实施例12:制备化合物va
[0153]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viic的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得22.7g化合物va,收率75%。非对映异构体比例7α:7β=7:1。
[0154]
ms(esi)m/z:477(m h
)。
[0155]1h nmr(400mhz,chloroform-d)δ5.82(s,1h),4.61(t,1h),3.52(t,2h),2.52(dd,j=14hz,j=2hz,1h),2.38~2.42(m,1h),2.21~2.29(m,3h),2.15~2.21(m,1h),2.03(s,3h),1.96~2.02(m,1h),1.85~1.88(m,1h),1.68~1.79(m,5h),1.46~1.60(m,4h),0.95~1.42(m,18h).
[0156]
实施例13:制备化合物va
[0157]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viid的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得11.0g化合物va,收率38%。
[0158]
实施例14:制备化合物va
[0159]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viie的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得10.0g化合物va,收率33%。
[0160]
实施例15:制备化合物va
[0161]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viif的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得8.0g化合物va,收率26%。
[0162]
实施例16:制备化合物va
[0163]
将化合物via(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viig的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得18.0g化合物va,收率60%。
[0164]
实施例17:制备化合物vb
[0165]
将化合物vib(2g)和氯化亚铜(263mg)加入反应瓶中,加入四氢呋喃(15ml),-30℃下,滴入化合物viia的四氢呋喃溶液(1.5eq.),滴加完毕后,反应继续在-30℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得2.24g化合物vb,收率78%。非对映异构体比例7α:7β=9:1。
[0166]
ms(esi)m/z:539(m h
)。
[0167]
实施例18:制备化合物vc
[0168]
将化合物vic(2g)和溴化亚铜(280mg)加入反应瓶中,加入四氢呋喃(15ml),0℃下,滴入化合物viia的四氢呋喃溶液(1.5eq.),滴加完毕后,反应继续在0℃搅拌1h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得2.13g化合物vc,收率70%。非对映异构体比例7α:7β=5:1。
[0169]
ms(esi)m/z:479(m h
)。
[0170]
实施例19:制备化合物vd
[0171]
将化合物vid(2g)和碘化亚铜(306mg)加入反应瓶中,加入四氢呋喃(15ml),-10℃下,滴入化合物viia的四氢呋喃溶液(1.2eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得2.35g化合物vd,收率81%。非对映异构体比例7α:7β=8:1。
[0172]
ms(esi)m/z:525(m h
)。
[0173]
实施例20:制备化合物ve
[0174]
将化合物vie(2g)和三氟甲磺酸亚铜(244mg)加入反应瓶中,加入2-甲基四氢呋喃(15ml),-30℃下,滴入化合物viib的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-30℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得2.28g化合物ve,收率80%。非对映异构体比例7α:7β=9:1。
[0175]
ms(esi)m/z:549(m h
)。
[0176]
实施例21:制备化合物vf
[0177]
将化合物vif(2g)和氯化亚铜(122mg)加入反应瓶中,加入四氢呋喃(15ml),-20℃下,滴入化合物viia的四氢呋喃溶液(1.3eq.),滴加完毕后,反应继续在-20℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得2.25g化合物vf,收率77%。非对映异构体比例7α:7β=9:1。
[0178]
ms(esi)m/z:519(m h
)。
[0179]
实施例22:制备化合物vg
[0180]
将化合物vig(20g)和氯化亚铜(1.42g)加入反应瓶中,加入四氢呋喃(120ml),-10℃下,滴入化合物viia的四氢呋喃溶液(2.5eq.),滴加完毕后,反应继续在-10℃搅拌2h,hplc显示原料转化完全。加入乙酸淬灭反应,加入水稀释,乙酸乙酯萃取,有机相用饱和碳酸氢钠洗涤,干燥后浓缩得18.6g化合物vg,收率58%。非对映异构体比例7α:7β=8:1。
[0181]
实施例23:制备化合物iiia
[0182]
将溴化铜(5.62g)和溴化锂(1.55g)加入反应瓶中,随后加入乙腈(15ml)和乙酸酐(1.23g),反应在20℃搅拌30min后,滴加入化合物va(5g)的乙腈(15ml)溶液,反应继续在20℃搅拌3h,hplc监控显示原料转化完全。将反应液滴加入硫脲的甲苯和水的混合液中,搅拌30min,加入碳酸氢钠,搅拌10min,垫硅藻土过滤,用甲苯洗涤,滤液分层后有机相用饱和氯化钠溶液洗涤,干燥后浓缩得4.87g化合物iiia,收率:90%。
[0183]
ms(esi)m/z:517(m h
)。
[0184]1h nmr(400mhz,chloroform-d)δ7.27(d,j=9.6hz,1h),6.87(dd,j=8.4hz,j=2.4hz,1h),6.78(d,j=2.4hz,1h),4.70(t,j=8.4hz,1h),3.53(t,j=6.8hz,2h),2.90(t,j=16.8hz,j=5.2hz,1h),2.75(d,j=16.8hz,1h),2.29~2.37(m,1h),2.28(s,3h),2.20~2.26(m,1h),2.06(s,3h),1.84~1.87(m,1h),1.72~1.79(m,3h),1.59~1.68(m,3h),1.37~1.55(m,8h),1.18~1.48(m,18h),0.82(s,1h)
[0185]
实施例24:制备化合物iiib
[0186]
将溴化铜(3.71g)和溴化锂(1.02g)加入反应瓶中,随后加入乙腈(10ml)和丙酸酐(0.77g),反应在20℃搅拌30min后,滴加入化合物va(3.3g)的乙腈(10ml)溶液,反应继续在20℃搅拌3h,hplc监控显示原料转化完全。将反应液滴加入硫脲的甲苯和水的混合液中,搅拌30min,加入碳酸氢钠,搅拌10min,垫硅藻土过滤,用甲苯洗涤,滤液分层后有机相用饱和氯化钠溶液洗涤,干燥后浓缩得3.24g化合物iiib,收率:88%。
[0187]
ms(esi)m/z:531(m h
)。
[0188]1h nmr(400mhz,chloroform-d)δ7.28(d,j=8.8hz,1h),6.84(dd,j=8.4hz,j=2.4hz,1h),6.78(d,j=2.4hz,1h),4.67(t,j=8.4hz,1h),3.52(t,j=6.8hz,2h),2.90(dd,j=16.8hz,j=5.2hz,1h),2.75(d,j=16.8hz,1h),2.56(q,j=15.2hz,2h),2.19~
2.39(m,3h),2.06(s,3h),1.83~1.87(m,1h),1.72~1.79(m,3h),1.62~1.67(m,1h),1.39~1.58(m,8h),1.17~1.27(m,14h),0.82(s,3h)
[0189]
实施例25:制备化合物ii
[0190]
将化合物iiia(4.38g)和化合物iv(4.5g)加入反应瓶中,加入dmf(25ml)。20℃下加入40%的氢氧化钠(8.4g)。反应继续在20℃搅拌2h,hplc显示原料转化完全。加入乙酸猝灭反应,加入水稀释,mtbe萃取,有机相依次用饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤,干燥后浓缩得到4.51g化合物ii,收率:90%。
[0191]
ms(esi)m/z:591(m h
)。
[0192]1h nmr(400mhz,chloroform-d)δ7.15(d,j=8.4hz,1h),6.63(dd,j=8.8hz,j=2.4hz,1h),6.55(d,j=2.4hz,1h),4.67(s,1h),3.75(t,j=7.6hz,1h),2.86(dd,j=16.8hz,j=5.2hz,1h),2.71(d,j=16.8hz,1h),2.59(t,j=6hz,2h),2.50(t,j=7.2hz,2h),2.28~2.34(m,2h),2.11~2.23(m,3h),1.84~1.92(m,3h),1.72~1.75(m,1h),1.53~1.65(m,4h),1.17~1.50(m,24h)
[0193]
实施例26:制备化合物ii
[0194]
将化合物iiib(3g)和化合物iv(3.08g)加入反应瓶中,加入dmf(20ml)。20℃下加入40%的氢氧化钠(5.25g)。反应继续在20℃搅拌2h,hplc显示原料转化完全。加入乙酸猝灭反应,加入水稀释,mtbe萃取,有机相依次用饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤,干燥后浓缩得到2.77g化合物ii,收率:83%。
[0195]
ms(esi)m/z:591(m h
)。
[0196]1h nmr(400mhz,chloroform-d)δ7.15(d,j=8.4hz,1h),6.63(dd,j=8.8hz,j=2.4hz,1h),6.55(d,j=2.4hz,1h),4.67(s,1h),3.75(t,j=7.6hz,1h),2.86(dd,j=16.8hz,j=5.2hz,1h),2.71(d,j=16.8hz,1h),2.59(t,j=6hz,2h),2.50(t,j=7.2hz,2h),2.28~2.34(m,2h),2.11~2.23(m,3h),1.84~1.92(m,3h),1.72~1.75(m,1h),1.53~1.65(m,4h),1.17~1.50(m,24h)
[0197]
实施例27:制备化合物i
[0198]
将化合物ii(4.31g)溶于乙酸乙酯(15ml)中,依次加入乙酸(2.63g)和17.5%的双氧水(3.12g),反应在20℃继续搅拌10h,hplc显示原料转化完全。加入亚硫酸钠溶液猝灭反应。加入乙酸乙酯萃取。有机相依次用饱和碳酸氢钠和氯化钠溶液洗涤,干燥后浓缩,乙酸乙酯重结晶得到3.52g化合物i,收率:80%。纯度:99.7%。
[0199]
ms(esi)m/z:607(m h
)。
[0200]1h nmr(400mhz,chloroform-d)δ7.11(d,j=8.4hz,1h),6.63(d,j=8.4hz,1h),6.55(s,1h),3.74(t,j=8.5hz,1h),2.90-2.53(m,6h),2.35-2.03(m,7h),1.99-1.83(m,1h),1.80-1.67(m,3h),1.60(qt,j=8.5,3.3hz,2h),1.53-1.11(m,19h),1.08-0.95(m,1h),0.77(s,3h).
[0201]
由于已根据其特殊的实施方案描述了本发明,某些修饰和等价变化对于本领域普通技术人员是显而易见的且包括在本发明的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。