氯吡嘧磺隆的gc-ms检测分析条件的建立方法
技术领域
1.本发明属于化学分析及检测领域,涉及氯吡嘧磺隆定性和定量的检测分析,具体涉及一种氯吡嘧磺隆的气相色谱-质谱(gc-ms)检测分析条件的建立方法。
背景技术:
2.氯吡嘧磺隆(halosulfuron-methyl),化学名称:3-(4,6-二甲氧基嘧啶-2-基)-1-(1-甲基-3-氯-4-甲氧基甲酰吡唑-5-基)磺酰脲,分子式c
13h15
c1n6o7s,分子量434.81,属于磺酰脲类除草剂。主要用于防除阔叶杂草和莎草科杂草,对玉米、水稻、小麦、甘蔗等禾本科作物具有很高的选择性。其作用机理为通过抑制植物的乙酰乳酸合成酶,阻止支链氨基酸等生物合成,最终破坏蛋白质的合成,干扰dna的合成及细胞的分裂和生长。
3.目前,国内外氯吡嘧磺隆的检测方法主要是液相色谱和液相色谱-质谱联用仪。还未见采用气相色谱-质谱法(gc-ms)测定氯吡嘧磺隆的研究报道。
技术实现要素:
4.本发明的目的是针对上述现有技术的不足,提供一种氯吡嘧磺隆的气相色谱-质谱法(gc-ms)检测分析条件的建立方法,以期通过建立的gc-ms检测分析条件对氯吡嘧磺隆进行定性和定量检测分析,该gc-ms检测分析方法具有快速、简便、灵敏度高、选择性强等优点。
5.为实现以上目的,本发明采用的技术方案是:一种氯吡嘧磺隆的gc-ms检测分析条件的建立方法,该方法步骤如下:
6.a.标准工作溶液的配制:取氯吡嘧磺隆标准品,利用丙酮溶解,再配制成标准工作溶液,使氯吡嘧磺隆质量浓度为0.01、0.05、0.10、0.50、1.00、5.0、10.0mg/l。
7.上述步骤中利用丙酮溶解氯吡嘧磺隆后是先配制质量浓度为1000.0mg/l的氯吡嘧磺隆标准母液,再将标准母液稀释成标准工作溶液。
8.b.全扫描模式检测确定定性定量离子:对配制氯吡嘧磺隆标准工作溶液的丙酮、1mg/l和10mg/l的氯吡嘧磺隆标准工作溶液进行全扫描模式检测,m/z50-600,得到氯吡嘧磺隆标准溶液的全扫气相色谱-质谱的总离子色谱图,以进一步得到氯吡嘧磺隆全扫质谱图,于该氯吡嘧磺隆全扫质谱图上找到灵敏度最大即丰度最大的3个特征离子碎片作为定性和定量离子,即m/z 139.10、260.10、327.10,其中定量离子为m/z 327.10;
9.所述氯吡嘧磺隆全扫质谱图的获得过程为:氯吡嘧磺隆标准溶液的全扫气相色谱-质谱的总离子色谱图中,除丙酮中出现的色谱峰外,若标准工作溶液中出现新的色谱峰且新出现的色谱峰的低浓度和高浓度成正比,则判断新出现的色谱峰是氯吡嘧磺隆的色谱峰;双击氯吡嘧磺隆的色谱峰,得到氯吡嘧磺隆全扫质谱图。
10.上述提及的全扫描模式的gc-ms条件为:色谱柱为安捷伦毛细管柱hp-5ms,30m
×
0.25mm
×
0.25um;进样口温度260℃,程序升温:60℃保持3.0min,20℃/min升温至290℃,保持10.0min;载气:氦气,纯度99.999%;不分流进样,进样量:1μl,流速1.0ml/min;四极杆温
度150℃,离子源温度230℃,质谱传输线温度280℃;溶剂延迟时间为2.0min,数据采集模式:全扫描检测,扫描m/z 50-600。
11.c.离子监测模式检测:在氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,定量离子为m/z 327.10的条件下,在gc-ms上采用离子监测模式检测标准工作溶液0.01、0.05、0.10、0.50、1.00mg/l,gc-ms其他检测条件同步骤b的全扫描gc-ms条件;若低浓度氯吡嘧磺隆的标准工作溶液的峰面积太小,不能积峰,有许多小杂峰出现,则灵敏度不够,需要调整仪器参数;
12.d.优化气相检测条件,提高氯吡嘧磺隆的灵敏度,确定氯吡嘧磺隆的gc-ms检测分析条件:在氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,定量离子为m/z 327.10的条件下,以步骤b的全扫描gc-ms条件为基础,通过改变变量进样量、进样口温度、流速、程序升温或离子源温度中的任一个变量的参数,在gc-ms上采用离子监测模式检测标准工作溶液1.00mg/l,以通过比较变量在不同参数检测条件下标准工作溶液的峰面积是否最大、出峰时间是否最合适、峰型是否对称,来确定氯吡嘧磺隆的gc-ms检测分析条件。
13.上述步骤d中确定的氯吡嘧磺隆的gc-ms检测分析条件为:色谱柱为安捷伦毛细管柱hp-5ms,30m
×
0.25mm
×
0.25um;进样口温度290℃,程序升温:130℃保持1.0min,10℃/min升温至250℃,保持1.0min;10℃/min升温至300℃,保持1.0min;载气:氦气,纯度99.999%;不分流进样,进样量:2μl,流速1.2ml/min;四极杆温度150℃,离子源温度230℃,质谱传输线温度280℃;溶剂延迟时间为2.0min,数据采集模式:离子监测模式,氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,其中定量离子为m/z 327.10。
14.以上述氯吡嘧磺隆的gc-ms检测分析条件检测氯吡嘧磺隆标准工作溶液0.01、0.05、0.10、0.50、1.00mg/l,得到的标准工作曲线方程为y=437997x-4409.8,r2=0.9995,是以氯吡嘧磺隆的质量浓度为横坐标、定量离子峰面积为纵坐标绘制而成。
15.本发明首次建立了检测氯吡嘧磺隆的gc-ms仪器分析条件,是通过查阅氯吡嘧磺隆的理化性质,大致设定gc-ms仪器参数,全扫找到氯吡嘧磺隆的定性和定量离子,同时对gc-ms检测参数(进样量,进样口温度,柱温箱温度等)进行优化,来提高分离效率和灵敏度。该gc-ms检测分析条件对应的gc-ms检测分析方法具有灵敏度高、精密度好、稳定性好等特点,分析的最低检测限为0.01mg/l,为氯吡嘧磺隆的检测提供一种新的检测方法。
附图说明
16.图1为氯吡嘧磺隆的全扫质谱图。
17.图2为实施例1中未优化气质条件前一系列氯吡嘧磺隆标准溶液的总离子色谱图。
18.图3为本发明1.00mg/l氯吡嘧磺隆标准工作溶液的离子监测气相色谱-质谱的总离子色谱图。
19.图4为本发明1.00mg/l氯吡嘧磺隆标准工作溶液的离子碎片相对丰度值图谱。
20.图5为本发明氯吡嘧磺隆的标准工作曲线图。
具体实施方式
21.下面的实施例是对本发明的进一步详细描述,但实施例并不局限于本发明的保护范围。
22.实施例所用仪器和试剂:
23.气相色谱-质谱仪(gc7890a-ms5975,安捷伦科技有限公司,美国),配有电子轰击电离离子源(ei)。色谱柱:agilent hp-5ms(30m
×
0.25mm
×
0.25um)。氯吡嘧磺隆标准品(纯度为97%),丙酮为色谱纯,购于天津迪博化工股份有限公司。
24.实施例1氯吡嘧磺隆的气相色谱-质谱仪检测条件的确认
25.1.标准工作溶液的配制
26.准确称取氯吡嘧磺隆标准品(纯度为97%)0.1031g(精确至0.0001g),置于100ml的棕色容量瓶中,用色谱丙酮溶解并配制成质量浓度为1000.0mg/l的氯吡嘧磺隆标准母液。然后采用梯度稀释法分别取氯吡嘧磺隆的标准母液配制成标准工作溶液,使氯吡嘧磺隆质量浓度为0.01、0.05、0.10、0.50、1.00、5.0、10.0mg/l。
27.2.全扫描模式检测找到定性定量离子
28.对配制氯吡嘧磺隆标准工作溶液的溶剂丙酮、1mg/l和10mg/l的氯吡嘧磺隆标准溶液进行全扫描(m/z50~600),得到氯吡嘧磺隆标准溶液的全扫气相色谱-质谱的总离子色谱图,扣除丙酮中出现的色谱峰,标准溶液中出现新的色谱峰且新出现的色谱峰的低浓度和高浓度成正比,判断新出现的色谱峰为氯吡嘧磺隆的色谱峰,双击该色谱峰,得到氯吡嘧磺隆全扫质谱图(图1),找到灵敏度最大即丰度最大的3个特征离子碎片作为定性和定量离子,即m/z 139.10、260.10、327.10,其中定量离子为m/z 327.10。
29.全扫描的gc-ms条件为:色谱柱为安捷伦毛细管柱hp-5ms,30m
×
0.25mm
×
0.25um;进样口温度260℃,程序升温:60℃保持3.0min,20℃/min升温至290℃,保持10.0min;载气:氦气,纯度99.999%;不分流进样,进样量:1μl,流速1.0ml/min;四极杆温度150℃,离子源温度230℃,质谱传输线温度280℃;溶剂延迟时间为2.0min,数据采集模式:全扫描检测,扫描m/z50-600。
30.3.离子监测模式检测
31.在氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,定量离子为m/z 327.10的条件下,在gc-ms上采用离子监测模式检测歩骤(1)配制好的标准工作溶液0.01、0.05、0.10、0.50、1.00mg/l,gc-ms其他检测条件同步骤2的全扫描gc-ms条件。参见图2,高浓度的标准溶液均在保留时间出峰,并且都成线性,同时都有氯吡嘧磺隆监测特征离子碎片。但低浓度标准溶液有出峰,但灵敏度不够,且最低浓度0.01因为灵敏度比较低,有许多小杂峰干扰,积分不准确,需要调整仪器参数。
32.4.优化气相检测条件,提高氯吡嘧磺隆的灵敏度
33.通过优化气相条件,通过改变进样量(1ul,1.5ul和2ul),进样口温度(260℃,270℃,280℃,290℃),流速(0.8ml/min,1.0ml/min,1.2ml/min);程序升温,离子源温度(210℃,220℃,230℃)等参数,来提高氯吡嘧磺隆的灵敏度。
34.在氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,定量离子为m/z 327.10的条件下,以步骤2的全扫描gc-ms检测条件为基础,通过改变上述变量进样量(1ul,1.5ul和2ul)、进样口温度(240℃,260℃,280℃,290℃)、流速(0.8ml/min,1.0ml/min,1.2ml/min)、程序升温或离子源温度(210℃,220℃,230℃)的不同参数,在gc-ms上采用离子监测模式检测步骤(1)配制的标准工作溶液1.00mg/l,以通过比较不同检测参数条件下标准工作溶液1.00mg/l的峰面积、出峰时间和峰型(峰面积越大越好,出峰时间是否合适,峰型是否对
称),来考查不同参数下的变量对氯吡嘧磺隆检测的影响,以确定检测氯吡嘧磺隆的gc-ms检测条件,结果参见下表1-表5。
35.表1进样量对氯吡嘧磺隆检测的影响
36.进样量(ul)峰面积出峰时间峰型121046113.983对称1.53206513.983对称2.041286113.983对称
37.进样量的选择需考虑仪器所用的色谱柱和称管,进样量过大很容易造成柱超载。本方法根据所选用的色谱柱和称管型号设定合理的不同进样量考察,由表1可知,进样量的变化对色谱峰型和出峰时间影响不大,但对响应峰面积的影响很大,2.0ul时峰面积最大,灵敏度最好,故确定进样量为2.0ul为检测条件。
38.表2进样口温度对氯吡嘧磺隆检测的影响
39.进样口温度(℃)峰面积出峰时间峰型24041031913.983对称26041049613.983对称28041198313.983对称29041286113.983对称
40.进样口温度的要求是能保证待测组分在瞬间不分流时汽化,但不能使待测组分分解。从表2可知,进样口温度的变化对色谱峰型和出峰时间影响不大,而且对响应峰面积的影响也不是很大,为了保证样品能够完全被气化,故确定选择最高温度290℃为检测条件。
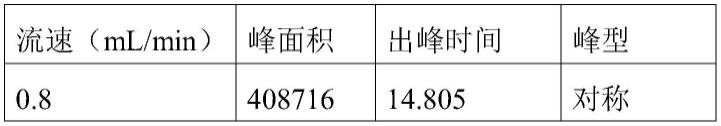
41.表3气体流速对氯吡嘧磺隆检测的影响
[0042][0043][0044]
在实际检测分析中,氦气流速要既能保证待测物的完全分离,又要保证峰面积、峰形达到检测的需求,并且分析时间越短越好。由表3可以看出,当流速为1.2ml/min时,峰面积最大、且峰形、分离度及重现性均较好,而且进样时间最短,故确定流动相流速为1.2ml/min为检测条件。
[0045]
表4程序升温对氯吡嘧磺隆检测的影响
[0046][0047]
通过改变炉温起始温度和不同升温速率组成不同升温程序对氯吡嘧磺隆的峰型、峰面积和出峰时间的影响来确定升温程序。从表4可知,程序升温的变化对色谱峰型和峰面积影响不大,但对出峰时间的影响很大,应用方法3的程序升温时峰面积最大,出峰时间最短,故确定程序升温为方法3为检测条件。
[0048]
表5离子源温度对氯吡嘧磺隆检测的影响
[0049]
离子源温度(℃)峰面积出峰时间峰型21040991213.983对称22040993813.983对称23041286113.983对称
[0050]
从表5可知,离子源温度的变化对色谱峰型、出峰时间以及峰面积的影响都不太大,但在230℃时,峰面积相对最大,灵敏度最好,故确定离子源温度为230℃为检测条件。
[0051]
因而在前述分析基础上,确定检测分析氯吡嘧磺隆的gc-ms条件为:色谱柱为安捷伦毛细管柱hp-5ms,30m
×
0.25mm
×
0.25um;进样口温度290℃,程序升温:130℃保持1.0min,10℃/min升温至250℃,保持1.0min;10℃/min升温至300℃,保持1.0min;载气:氦气,纯度99.999%;不分流进样,进样量:2μl,流速1.2ml/min;四极杆温度150℃,离子源温度230℃,质谱传输线温度280℃;溶剂延迟时间为2.0min,数据采集模式:离子监测模式,氯吡嘧磺隆监测离子m/z 139.10、260.10、327.10,其中定量离子为m/z 327.10。
[0052]
5.定性和定量分析
[0053]
定性分析:在上述确定的gc-ms条件下检测氯吡嘧磺隆标准工作溶液0.01、0.05、0.10、0.50、1.00mg/l。结果显示,不同梯度浓度的氯吡嘧磺隆标准工作溶液的气相色谱-质谱的总离子色谱图中的氯吡嘧磺隆的出峰时间基本一致,图3显示了1.00mg/l氯吡嘧磺隆标准工作溶液的离子监测气相色谱-质谱的总离子色谱图,可知氯吡嘧磺隆标准溶液在13.983分钟保留。不同梯度浓度的氯吡嘧磺隆标准工作溶液的离子碎片相对丰度值图谱相同,图4显示了1.00mg/l氯吡嘧磺隆标准工作溶液的离子碎片相对丰度值图谱,由图可知其监测离子为m/z139.10、260.10、327.10。
[0054]
定量分析:采用气相色谱-质谱结合外标法进行。将前述配制的氯吡嘧磺隆标准工作溶液0.01、0.05、0.10、0.50、1.00mg/l,在上述确定的gc-ms检测条件下进行测定,每个质量浓度进样3次,共进5个浓度。以氯吡嘧磺隆的质量浓度(x,mg/l)为横坐标、定量离子峰面积(y)为纵坐标进行性回归,并计算相关系数(r)。结果见表6和图5。
[0055]
表6氯吡嘧磺隆的标准工作曲线
[0056][0057][0058]
从表6和图5可知,氯吡嘧磺隆的标准工作曲线方程为y=437997x-4409.8,r2=0.9995,线性关系良好。按上述方法分析的最低检测限为0.01mg/l。
[0059]
实施例2氯吡嘧磺隆的气相色谱-质谱仪检测方法的线性关系、精密度和准确度考察
[0060]
1.线性关系考察
[0061]
分别移取氯吡嘧磺隆标准工作溶液贮备液适量,用丙酮配制成氯吡嘧磺隆质量浓度分别为0.01、0.05、0.10、0.50、1.00mg/l的系列对照品标准溶液。放入进样小瓶中,放入自动进样器中,运行自动进样程序注入gc-ms分析仪,检测分析条件见实施例1确定的条件,每个质量浓度进样3次,共进5个点。以目标化合物的质量浓度(x,mg/l)为横坐标、定量离子峰面积(y)为纵坐标进行回归分析,并计算相关系数(r)。结果见表6和图5。由表6和图5可知,氯吡嘧磺在一定线性范围内的峰面积与浓度呈良好的线性关系,相关系数为0.9995。
[0062]
检测限(lod)是在样品测量过程中,在相同条件下,分析方法可以检测到的分析对象的最低浓度,本方明确定的gc-ms检测条件分析的氯吡嘧磺隆的最低检测限为0.01mg/l。
[0063]
2.分析方法精密度测定
[0064]
取已配制好的氯吡嘧磺隆的标准溶液(1mg/l),分装到5个进样瓶中。称取一定量的75%氯吡嘧磺隆水分散颗粒剂,置于100ml的容量瓶中,用色谱丙酮溶解并稀释至刻度,摇匀,形成试样溶液。用同样的方法称取5份平行试样,形成5份试样溶液,放入进样小瓶中,然后放入自动进样器中,按着先放标准溶液,然后试样溶液的顺序运行自动进样程序注入gc-ms分析仪中,按照实例例1确定的gc-ms检测分析条件进样检测(试样中氯吡嘧磺隆的质量分数计算为现有常规技术),检验试样测定精密度,结果见表7。实验结果表明:5份氯吡嘧磺隆农药试样含量的标准偏差为0.02,变异系数为2.48%,样品测定方法精密度良好。
[0065]
表7氯吡嘧磺隆gc-ms方法的精密度试验结果
[0066][0067]
3.分析方法的准确度测定
[0068]
为了考察分析方法的准确度,对试样进行加标回收实验。在已知含量的75%氯吡嘧磺隆水分散颗粒剂试样中分别加入一定量的氯吡嘧磺隆标样,用实施例1确定的gc-ms检测分析方法进行测定其含量,并计算加标回收率(回收率计算为现有常规技术)。结果见表
8,结果表明,氯吡嘧磺的平均回收率分别为103%。方法准确度较高,可满足定量分析要求。
[0069]
表8氯吡嘧磺隆方法的准确度试验结果
[0070][0071]
由上可知,本发明建立的gc-ms检测分析条件对应的gc-ms检测分析方法的准确度和精密度较高,线性关系良好,具有简便、快速、准确级分离效果好的优点,是一种可行的分析方法。
[0072]
以上实施例仅用以说明本发明的技术方案,而非对其限制,其目的在于让熟悉此项技术的人员能够了解本发明的内容并据以实施,凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。