一种氰胺类化合物的一锅法制备工艺
(一)技术领域
1.本发明涉及氰胺类化合物的制备工艺,特别涉及一种利用羟胺作为氨源,氟磺酸咪唑盐作为促进剂,使腈类化合物发生羟胺化反应、蒂曼重排反应生成氰胺类化合物的一锅法制备工艺。
(二)
背景技术:
2.氰胺类化合物作为一种同时具有氨基和氰基双官能团的化学物种,是一类重要的二氮类化合物,具有多种生物活性,被广泛地应用于医药、农药以及材料科学等领域。由于其特殊的n-c-n键结构,氰胺类化合物是一种非常重要的精细化工原料和有机合成领域的关键中间体,同时也是一种配位效果极好的金属配体。介于氰胺类化合物的广泛应用前景,自二十世纪以来,化学家对氰胺的合成方法及其在有机合成领域中的应用进行了系统全面的研究。制备氰胺类化合物最传统的方法之一是采用卤化氰对胺直接进行氰基化反应而获得。从反应过程来看,高毒性卤化氰的使用限制了此方法的大规模应用。近年来,关于以低毒性的化合物为原料发展的氰胺合成策略的报道越来越多,如satyanarayan等在2009年报道了以氨基二硫甲酸的铵盐为原料,串联三步反应直接合成了氰胺的一锅法策略[green chem.,2009,11,1503.];陈教授课题组于2014年报道了以低毒性的三甲基氰硅烷(tmscn)为氰化试剂即氮源,通过次氯酸钠/水促进仲胺的亲电取代氰化反应合成二取代氰胺的方法[org.lett.,2014,16,247.];chien等报道了苯磺酰氯化物(tscl或o-nscl)促进脒肟的蒂曼重排反应,生成相应的氰胺类化合物[org.lett.,2014,16,892.]。然而,以上方法都存在一些缺点,如需要高温、高压等苛刻的反应条件,需要添加化学计量的化学试剂,体系对环境不友好,不利于大规模应用。
[0003]
氟磺酰咪唑盐的结构如a所示,作为一种稳定、易存储运输的固体类化合物,由董佳家教授等人首次报道合成,并将其作为高效的磺酰氟试剂,在室温和碱条件下即可完成对苯酚、杂芳基酚的氟磺酰化。该反应表现出了非常好的兼容性和效率,且不需过渡金属参与等优点[angew.chem.,int.ed.,2018,57,2605.]。
[0004][0005]
现有方法中未有采用氟磺酰咪唑盐制备氰胺类化合物的报道,需要寻找一种高效、环保、经济的合成氰胺类化合物的合成工艺。
(三)
技术实现要素:
[0006]
本发明针对现有技术中存在的缺陷,提供一种高效、环保、经济的合成氰胺类化合物的一锅法制备新工艺,本发明采用价廉易得的羟胺作为氨源,以稳定、易储存的氟磺酰咪唑盐作为促进剂,以常见的腈类化合物为底物,经一锅法先后发生羟胺化反应、蒂曼重排反应合成氰胺类化合物,收率得到了显著提高。
[0007]
本发明采用的技术方案是:
[0008]
本发明提供一种氰胺类化合物的一锅法制备工艺,所述制备工艺包括如下步骤:
[0009]
以式(i)所示腈类化合物为原料,先加入羟胺、溶剂,于回流温度下反应1~5h后,反应液冷却至室温,加入式a所示氟磺酰咪唑盐、碱,于25~50℃下反应1~5h,反应液分离纯化,制得(ⅱ)所示的氰胺类化合物;所述碱为下列之一:碳酸钠(na2co3)、碳酸钾(k2co3)、碳酸氢钠(nahco3)、碳酸氢钾(khco3)、1,8-二氮杂二环十一碳-7-烯(dbu)、三乙胺(et3n)或二异丙基乙胺(dipea);所述溶剂为下列之一:水、乙醇、甲醇、乙腈、二氯甲烷、乙酸乙酯、甲苯、四氢呋喃或二甲基甲酰胺;
[0010][0011]
式(i)中r为芳香基、c1-c10直链或支链烷基,式(ii)中r与式(i)中r相同。
[0012]
进一步,式(i)中r为苯基、4-联苯基、对溴苯基(p-brph)、对三氟甲基苯基(p-cf3ph)或3-甲基苯基。
[0013]
进一步,所述羟胺以质量浓度50%羟胺水溶液形式加入,所述羟胺与式(i)所示腈类化合物的物质的量之比为1~15:1,优选5-10:1,更优选6-7:1。所述溶剂体积用量以式(i)所示腈类化合物物质的量计为1~30ml/mmol,优选2ml/mmol。所述氟磺酰咪唑盐与式(i)所示腈类化合物的物质的量之比为1~5:1,优选1-3:1。所述碱与式(i)所示腈类化合物的物质的量之比为0.1~5:1,优选0.1-0.3:1。
[0014]
进一步,所述回流温度下反应时间为3h。
[0015]
进一步,所述反应温度优选为25~30℃,反应时间为2h。
[0016]
进一步,所述反应液分离纯化的方法为:反应结束后,反应液用体积比1:1的乙酸乙酯/乙醚溶液进行重结晶,取晶体,得到(ⅱ)所示的氰胺类化合物;所述乙酸乙酯/乙醚溶液体积用量以式(i)所示腈类化合物物质的量计为0.1~3ml/mmol,优选0.2ml/mmol。
[0017]
与现有技术相比,本发明的有益效果主要体现在:
[0018]
1、本发明使用价廉易得、环境友好的羟胺作为氨源,高效促进腈进行羟胺化,生成脒肟中间体。
[0019]
2、本发明使用结构稳定、易于储存的固体氟磺酰咪唑盐(a)作为促进剂,高效促进脒肟中间体进行蒂曼重排,生成氰胺。
[0020]
3、羟胺、氟磺酰咪唑盐(a)避免了使用氰化物等危险试剂,因此可以作为腈一锅法制备氰胺的标准处理条件的绿色替代品。
[0021]
4、最终产物中的氮仅来自于底物腈和羟胺,无需额外的胺加入。
[0022]
5、操作过程简单,后处理只需通过重结晶即可得到,产物收率高达95%,适合大规模制备。
(四)附图说明
[0023]
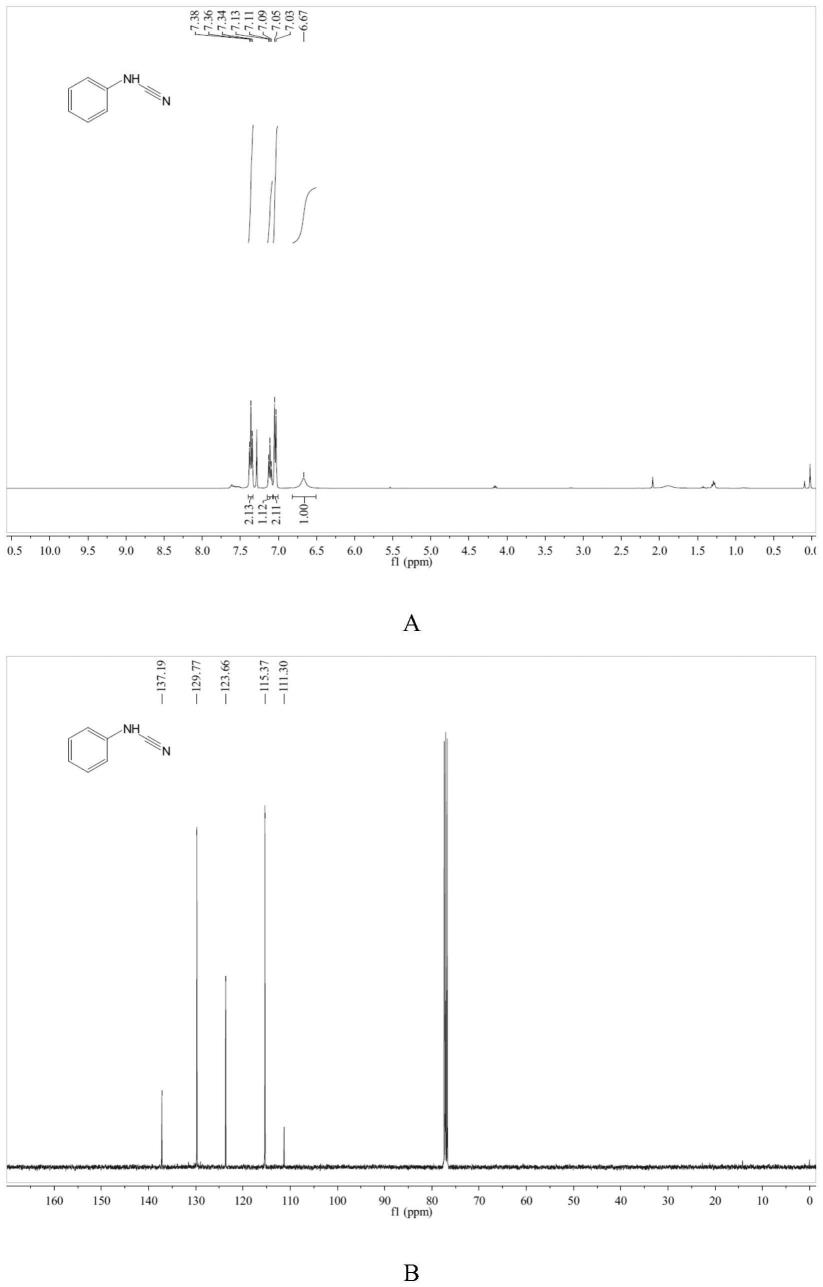
图1为实施例1制备化合物的核磁共振氢谱(a)、碳谱(b)图。
[0024]
图2为实施例2制备化合物的核磁共振氢谱(a)、碳谱(b)图。
[0025]
图3为实施例3制备化合物的核磁共振氢谱(a)、碳谱(b)图。
[0026]
图4为实施例4制备化合物的核磁共振氢谱(a)、碳谱(b)图。
[0027]
图5为实施例5制备化合物的核磁共振氢谱(a)、碳谱(b)图。
(五)具体实施方式
[0028]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:本发明所述室温为25-30℃。
[0029]
实施例1:n-苯基氰胺的制备
[0030]
在一个100ml单口烧瓶中,依次加入苯甲腈(式
ⅰ‑
1,r=ph)1.03g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入4.0g(12.2mmol)氟磺酰咪唑盐(a),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h;反应结束后,反应液用2ml体积比1:1的乙酸乙酯/乙醚溶液进行重结晶,取晶体,得到n-苯基氰胺(式
ⅱ‑
1,r=ph)1.12g,核磁共振氢谱图见图1中a所示,核磁共振碳谱图见图1中b所示,收率95%。
[0031]
核磁共振氢谱:(400mhz,chloroform-d)δ7.36(t,j=7.9hz,2h),7.11(t,j=7.4hz,1h),7.04(d,j=7.8hz,2h),6.67(s,1h).。
[0032]
核磁共振碳谱:(101mhz,chloroform-d)δ137.19,129.77,123.66,115.37,111.30。
[0033][0034]
对照例1:n-苯基氰胺的制备
[0035]
在一个100ml单口烧瓶中,依次加入苯甲腈(式
ⅰ‑
1,r=ph)1.03g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入3.0g(12.2mmol)咪唑盐(a’),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h,无反应。
[0036][0037]
实施例2:n-([1,1'-联苯]-4-基)氰胺的制备
[0038]
在一个50ml单口烧瓶中,依次加入[1,1'-联苯]-4-基腈(式
ⅰ‑
2,r=4-联苯基)1.79g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入4.0g(12.2mmol)氟磺酰咪唑盐(a),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h;反应结束后,反应液用2ml体积比1:1的乙酸乙酯/乙醚溶液进行重结晶,取晶体,得到n-([1,1'-联苯]-4-基)氰胺(式
ⅱ‑
2,r=4-联苯基)1.65g,核磁共振氢谱图见图2中a所示,核磁共振碳谱图见图2中b所示,收率85%。
[0039]
核磁共振氢谱:(400mhz,dmso-d6)δ10.31(s,1h),7.70
–
7.65(m,2h),7.64
–
7.60(m,2h),7.45(t,j=7.7hz,2h),7.34(t,j=7.3hz,1h),7.06(dd,j=9.0,2.1hz,2h)。
[0040]
核磁共振碳谱:(101mhz,dmso-d6)δ139.87,138.54,134.98,129.41,128.51,127.58,126.70,115.91,112.46。
[0041]
[0042]
实施例3:n-(p-溴)苯基氰胺的制备
[0043]
在一个50ml单口烧瓶中,依次加入对溴苯甲腈(式
ⅰ‑
3,r=p-brph)1.82g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入4.0g(12.2mmol)氟磺酰咪唑盐(a),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h;反应结束后,反应液用乙酸乙酯/乙醚溶液进行重结晶,即可得到n-(p-溴)苯基氰胺(式
ⅱ‑
3,r=p-brph)1.76g,核磁共振氢谱图见图3中a所示,核磁共振碳谱图见图3中b所示,收率90%。
[0044]
核磁共振氢谱:(400mhz,dmso-d6)δ10.37(s,1h),7.58
–
7.42(m,2h),6.99
–
6.85(m,2h)。
[0045]
核磁共振碳谱:(101mhz,dmso-d6)δ138.62,132.93,117.53,114.50,112.05。
[0046][0047]
实施例4:n-(4-三氟甲基)苯基氰胺的制备
[0048]
在一个50ml单口烧瓶中,依次加入4-(三氟甲基)苯甲腈(式
ⅰ‑
4,r=p-cf3ph)1.71g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入4.0g(12.2mmol)氟磺酰咪唑盐(a),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h;反应结束后,反应液用2ml体积比1:1的乙酸乙酯/乙醚溶液进行重结晶,取晶体,得到n-(4-三氟甲基)苯基氰胺(式
ⅱ‑
4,r=p-cf3ph)1.45g,核磁共振氢谱图见图4中a所示,核磁共振碳谱图见图4中b所示,收率78%。
[0049]
核磁共振氢谱:(400mhz,chloroform-d)δ7.64(d,j=8.4hz,2h),7.14(d,j=8.4hz,2h),6.83(s,1h)。
[0050]
核磁共振碳谱:(101mhz,chloroform-d)δ140.29,127.27,127.23,127.20,127.16,126.27,125.94,125.21,115.36,110.05。
[0051][0052]
实施例5:n-(m-甲基)苯基氰胺的制备
[0053]
在一个50ml单口烧瓶中,依次加入3-甲基苯腈(式
ⅰ‑
5,r=3-甲基苯基)1.17g(10mmol),20ml乙腈,质量浓度50wt%羟胺水溶液(含2.0g羟胺,60.6mmol),升至回流温度下搅拌反应3.0h;反应液冷却至室温,加入4.0g(12.2mmol)氟磺酰咪唑盐(a),300μl(2.1mmol)三乙胺,25℃条件下搅拌3.0h;反应结束后,反应液用2ml体积比1:1的乙酸乙酯/乙醚溶液进行重结晶,取晶体,得到n-苄基氰胺(式
ⅱ‑
5,r=3-甲基苯基)1.04g,核磁共振氢谱图见图5中a所示,核磁共振碳谱图见图5中b所示,收率79%。
[0054]
核磁共振氢谱:(400mhz,chloroform-d)δ7.24(t,j=7.8hz,1h),6.92(d,j=7.6hz,1h),6.89
–
6.79(m,2h),6.41(s,1h),2.36(s,3h)。
[0055]
核磁共振碳谱:(101mhz,chloroform-d)δ140.04,137.04,129.58,124.53,115.94,112.49,111.33,21.42。
[0056]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。