1.本发明属于医药化工技术领域,尤其涉及一种甲磺酸仑伐替尼杂质及其制备方法。
背景技术:
2.乐伐替尼(lenvatinib),商品名lenvima,别名仑伐替尼,是由日本卫材(eisa)公司开发的一种口服多靶点受体酪氨酸激酶(rtk)抑制剂。据报道,该类药物可靶向作用于血管内皮生长因子受体(vegfr)1-3、成纤维细胞生长因子受体(fgfr)1-3、干细胞生长因子受体和β型血小板衍生生长因子受体(pdgfr),用于治疗局部复发或转移性、进展性、放射性碘难治性分化型甲状腺癌(dtc),也适用于肾细胞癌(rcc)、神经胶质瘤、肝细胞癌(hcc)等多种癌症的治疗。甲磺酸乐伐替尼类药物在多年的临床治疗的实践中取得了很多突破性成果,得到了全世界范围内医生和患者的广泛认可。
3.甲磺酸仑伐替尼杂质,对甲磺酸仑伐替尼的药品质量控制有重要的作用,因此制备甲磺酸仑伐替尼杂质标准品,对甲磺酸仑伐替尼的临床、药理、药代动力学、毒理等方面的分析研究,具有非常重要的意义,可以为全面分析甲磺酸仑伐替尼提供基准物质。
4.有鉴于此,特提出本发明。
技术实现要素:
5.本发明的目的在于提供一种甲磺酸仑伐替尼杂质及其制备方法,以解决上述问题。
6.为实现以上目的,本发明特采用以下技术方案:
7.一种甲磺酸仑伐替尼杂质,其结构式为:
[0008][0009]
其中,r为h或ch3。
[0010]
一种所述的甲磺酸仑伐替尼杂质的制备方法,包括:
[0011]
将4-氯-7-甲氧基喹啉-6-酰胺、钠盐和碘甲烷依次溶于n,n-二甲基甲酰胺中并进行恒温反应,恒温反应完成后,向体系中加冰水淬灭反应,然后依次进行洗涤、萃取、干燥和纯化,得到中间体化合物a和/或化合物b;
[0012]
向4-氨基-3-氯苯酚盐酸盐溶液中加入碱,一次升温后进行搅拌,然后加入中间体化合物a和/或化合物b,进行二次升温并反应,反应结束后依次进行冷却、加冷水淬灭、洗涤、萃取、干燥和纯化,即得甲磺酸仑伐替尼杂质。
[0013]
可选的,所述4-氯-7-甲氧基喹啉-6-酰胺和所述碘甲烷的摩尔质量比为1:1-6;
[0014]
优选地,所述钠盐为氢化钠和/或碳酸钠。
[0015]
可选的,所述恒温反应的温度为0-25℃,时间为8-15h。
[0016]
可选的,所述洗涤采用饱和食盐水进行;
[0017]
优选地,所述萃取使用二氯甲烷进行;
[0018]
优选地,所述干燥包括采用无水硫酸钠,将有机层旋干;
[0019]
优选地,所述纯化为经硅胶柱色谱纯化。
[0020]
可选的,所述中间体化合物a和化合物b的结构式为
[0021][0022]
其中,r为h或ch3。
[0023]
可选的,所述4-氨基-3-氯苯酚盐酸盐、碱和化合物a或化合物b的摩尔质量比为1:2-5:0.5-1。
[0024]
可选的,所述4-氨基-3-氯苯酚盐酸盐溶液的溶剂可以为n,n-二甲基甲酰胺和/或二甲基亚砜;
[0025]
优选地,所述碱可以为碳酸铯、氢氧化钾、叔丁醇钾和碳酸钾中的一种或多种。
[0026]
可选的,所述一次升温的目标温度为40-90℃,所述搅拌的时间为1-2h。
[0027]
可选的,所述二次升温的目标温度为100-130℃,所述反应的时间为8-15h。
[0028]
本发明的有益效果:
[0029]
本发明同时提供两种新的甲磺酸仑伐替尼杂质,对甲磺酸仑伐替尼的临床、药理、药代动力学、毒理等方面的分析研究具有重要意义。
[0030]
本发明创造性地通过4-氯-7-甲氧基喹啉-6-酰胺一步合成中间体化合物a和b,再分别通过中间体化合物a和b为底物,直接合成两种甲磺酸仑伐替尼杂质,不需要对原料基团进行保护,减少了反应步骤,节约时间,且所采用的试剂普遍易得,反应条件温和,反应产率高,合成制备成本较低,有利于工业化扩大生产。
附图说明
[0031]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0032]
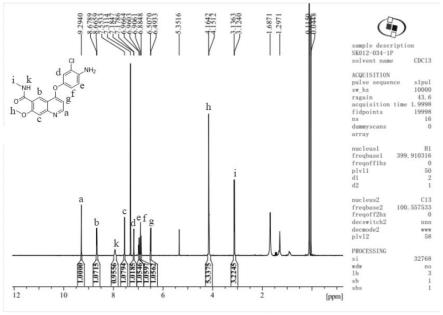
图1为实施例1所制备的甲磺酸仑伐替尼杂质m1z14的hnmr图谱;
[0033]
图2为实施例1所制备的甲磺酸仑伐替尼杂质m1z14的质谱图谱;
[0034]
图3为实施例1所制备的甲磺酸仑伐替尼杂质m1z14的液相图谱;
[0035]
图4为实施例1所制备的甲磺酸仑伐替尼杂质m1z15的hnmr图谱;
[0036]
图5为实施例1所制备的甲磺酸仑伐替尼杂质m1z15的质谱图谱;
[0037]
图6为实施例1所制备的甲磺酸仑伐替尼杂质m1z15的液相图谱。
具体实施方式
[0038]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例只是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0039]
首先,对本发明进行整体性解释,具体如下:
[0040]
本发明提供一种甲磺酸仑伐替尼杂质,其结构式为:
[0041][0042]
其中,r为h或ch3。
[0043]
当r为h时,所述的甲磺酸仑伐替尼杂质结构式为当r为ch3时,所述的甲磺酸仑伐替尼杂质结构式为
[0044]
本发明提供的新的甲磺酸仑伐替尼杂质对于甲磺酸仑伐替尼的药品质量控制有重要的作用,对甲磺酸仑伐替尼的临床、药理、药代动力学、毒理等方面的分析研究具有非常重要的意义;同时,该甲磺酸仑伐替尼杂质分子的合成对于其他仑伐替尼杂质的合成以及甲磺酸仑伐替尼药品的合成研究具有借鉴作用。
[0045]
本发明还提供一种所述的甲磺酸仑伐替尼杂质的制备方法,包括:
[0046]
将4-氯-7-甲氧基喹啉-6-酰胺、钠盐和碘甲烷依次溶于n,n-二甲基甲酰胺中并进行恒温反应,恒温反应完成后,向体系中加冰水淬灭反应,然后依次进行洗涤、萃取、干燥和纯化,得到中间体化合物a和/或化合物b;
[0047]
向4-氨基-3-氯苯酚盐酸盐溶液中加入碱,一次升温后进行搅拌,然后加入中间体化合物a和/或化合物b,进行二次升温并反应,反应结束后依次进行冷却、加冷水淬灭、洗涤、萃取、干燥和纯化,即得甲磺酸仑伐替尼杂质。
[0048]
在一个可选的实施方式中,所述4-氯-7-甲氧基喹啉-6-酰胺和所述碘甲烷的摩尔质量比为1:1-6;
[0049]
优选地,所述钠盐为氢化钠和/或碳酸钠。
[0050]
可选的,所述4-氯-7-甲氧基喹啉-6-酰胺和所述碘甲烷的摩尔质量比可以为1:1、1:2、1:3、1:4、1:5以及1:6之间的任意值。
[0051]
在一个可选的实施方式中,所述恒温反应的温度为0-25℃,时间为8-15h。
[0052]
可选的,所述恒温反应的温度可以为0℃、5℃、10℃、15℃、20℃以及25℃之间的任意值,时间可以为8h、9h、10h、11h、12h、13h、14h以及15h之间的任意值。
[0053]
在一个可选的实施方式中,所述洗涤采用饱和食盐水进行;
[0054]
优选地,所述萃取使用二氯甲烷进行;
[0055]
优选地,所述干燥包括采用无水硫酸钠,将有机层旋干;
[0056]
优选地,所述纯化为经硅胶柱色谱纯化。
[0057]
在一个可选的实施方式中,所述中间体化合物a和化合物b的结构式为
[0058][0059]
其中,r为h或ch3。
[0060]
当r为h时,中间体为化合物a,其结构式为当r为ch3时,中间体为化合物b,其结构式为
[0061]
在一个可选的实施方式中,所述4-氨基-3-氯苯酚盐酸盐、碱和化合物a或化合物b的摩尔质量比为1:2-5:0.5-1。
[0062]
可选的,所述4-氨基-3-氯苯酚盐酸盐、碱和化合物a或化合物b的摩尔质量比为1:2:0.5、1:3:0.6、1:4:0.7、1:5:0.8、1:3:0.9、1:2:1、1:5:0.5以及1:3:0.5之间的任意值。
[0063]
在一个可选的实施方式中,所述4-氨基-3-氯苯酚盐酸盐溶液的溶剂可以为n,n-二甲基甲酰胺和/或二甲基亚砜;
[0064]
优选地,所述碱可以为碳酸铯、氢氧化钾、叔丁醇钾和碳酸钾中的一种或多种。
[0065]
在一个可选的实施方式中,所述一次升温的目标温度为40-90℃,所述搅拌的时间为1-2h。
[0066]
可选的,所述一次升温的目标温度可以为40℃、50℃、60℃、70℃、80℃以及90℃之间的任意值,所述搅拌的时间可以为1h、1.1h、1.2h、1.3h、1.4h、1.5h、1.6h、1.7h、1.8h、1.9h以及2h之间的任意值。
[0067]
在一个可选的实施方式中,所述二次升温的目标温度为100-130℃,所述反应的时间为8-15h。
[0068]
可选的,所述二次升温的目标温度可以为100℃、105℃、110℃、115℃、120℃、125℃以及130℃之间的任意值,所述反应的时间可以为8h、9h、10h、11h、12h、13h、14h以及15h之间的任意值。
[0069]
本发明提供的制备方法其合成路径如下:
[0070][0071]
本发明创造性地通过4-氯-7-甲氧基喹啉-6-酰胺一步合成两种中间体化合物a和b,再分别通过中间体化合物a和b为底物,直接合成两种甲磺酸仑伐替尼杂质,不需要对原料基团进行保护,且原料价格便宜,简单易得,保存方便,反应简单高效,适用于工业化扩大生产的特点。
[0072]
实施例1
[0073]
使用本发明提供的甲磺酸仑伐替尼杂质的制备方法制备甲磺酸仑伐替尼杂质:
[0074]
s1:制备中间体化合物a和b:
[0075]
取5g21mmol的4-氯-7-甲氧基喹啉-6-酰胺加入到干燥反应瓶中,加入65ml的n,n-二甲基甲酰胺,缓慢加入氢化钠固体和34mmol的碘甲烷4.8g,控制温度为5℃,恒温反应15h。反应完成后加入少量冰水淬灭反应,用二氯甲烷萃取并收集有机相,将有机相浓缩硅胶柱色谱纯化即得到浅黄色化合物a和b。经计算,产率为84%;
[0076]
s2:制备甲磺酸仑伐替尼杂质:
[0077]
1.依次取13mmol的4-氨基-3-氯苯酚盐酸盐2.3g、n,n-二甲基甲酰胺30ml和52mmol的碳酸钾7.2g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入2g 8mmol的化合物a,在氮气条件下升温120℃反应15小时。反应完成后加入少量冷水淬灭反应,用二氯甲烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n-甲基喹啉-6-羧酸,命名为m1z14,其结构式为其hnmr图谱如图1所示,质谱图谱如图2所示,液相图谱如图3所示。
[0078]
经计算,产物质量2.34g,产率为82%。
[0079]
2.依次取13mmol的4-氨基-3-氯苯酚盐酸盐2.3g、n,n-二甲基甲酰胺30ml和52mmol的碳酸钾7.2g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入2g 7.5mmol的化合物b,在氮气条件下升温120℃反应15小时。反应完成后加入少量冷水淬灭反应,用二氯甲
烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n,n-二甲基喹啉-6-羧酸,命名为m1z15,其结构式为其hnmr图谱如图4所示,质谱图谱如图5所示,液相图谱如图6所示。
[0080]
经计算,产物质量为2.44g,产率为87%。
[0081]
实施例2
[0082]
使用本发明提供的甲磺酸仑伐替尼杂质的制备方法制备甲磺酸仑伐替尼杂质:
[0083]
s1:制备中间体化合物a和b:
[0084]
取1g4.2mmol的4-氯-7-甲氧基喹啉-6-酰胺加入到干燥反应瓶中,加入12ml的n,n-二甲基甲酰胺,缓慢加入氢化钠固体和7mmol的碘甲烷1g,控制温度为5℃,恒温反应12h。反应完成后加入少量冰水淬灭反应,用二氯甲烷萃取并收集有机相,将有机相浓缩硅胶柱色谱纯化即得到浅黄色化合物a和b。经计算,产率为85%;
[0085]
s2:制备甲磺酸仑伐替尼杂质:
[0086]
1.依次取2.8mmol的4-氨基-3-氯苯酚盐酸盐0.5g、n,n-二甲基甲酰胺5ml和12.3mmol的碳酸钾1.7g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入0.5g 2mmol的化合物a,在氮气条件下升温120℃反应15小时。反应完成后加入少量冷水淬灭反应,用二氯甲烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n-甲基喹啉-6-羧酸,命名为m1z14,其结构式为
[0087]
经计算,产物质量0.64g,产率为90%。
[0088]
2.依次取2.8mmol的4-氨基-3-氯苯酚盐酸盐0.5g、n,n-二甲基甲酰胺5ml和12.3mmol的碳酸钾1.7g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入0.52g 2mmol的化合物b,在氮气条件下升温120℃反应15小时。反应完成后加入少量冷水淬灭反应,用二氯甲烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n,n-二甲基喹啉-6-羧酸,命名为m1z15,其结构式为
[0089]
经计算,产物质量为0.68g,产率为92%。
[0090]
实施例3
[0091]
使用本发明提供的甲磺酸仑伐替尼杂质的制备方法制备甲磺酸仑伐替尼杂质:
[0092]
s1:制备中间体化合物a和b:
[0093]
取0.2g 0.84mmol的4-氯-7-甲氧基喹啉-6-酰胺加入到干燥反应瓶中,加入3ml的n,n-二甲基甲酰胺,缓慢加入氢化钠固体和1.4mmol的碘甲烷0.2g,反应8h。反应完成后加入少量冰水淬灭反应,用二氯甲烷萃取并收集有机相,将有机相浓缩硅胶柱色谱纯化即得到浅黄色化合物a和b。
[0094]
经计算,产率为85%;
[0095]
s2:制备甲磺酸仑伐替尼杂质:
[0096]
1.依次取0.44mmol的4-氨基-3-氯苯酚盐酸盐0.08g、n,n-二甲基甲酰胺1ml和1.81mmol的碳酸钾0.25g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入0.066g 0.26mmol的化合物a,在氮气条件下升温120℃反应15小时。反应完成后加入少量冷水淬灭反应,用二氯甲烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n-甲基喹啉-6-羧酸,命名为m1z14,其结构式为
[0097]
经计算,产物质量0.081g,产率为87%。
[0098]
2.依次取0.53mmol的4-氨基-3-氯苯酚盐酸盐0.095g、n,n-二甲基甲酰胺1.2ml和2.17mmol的碳酸钾0.3g,加入到反应瓶中,升温至80℃反应搅拌1小时,加入0.098g 0.37mmol的化合物b,在氮气条件下升温120℃反应15小时。反应完成后加入少量水淬灭反应,用二氯甲烷萃取有机相,饱和食盐水洗涤有机层三次,收集有机相,用无水硫酸钠干燥,减压蒸馏浓缩旋干,硅胶柱色谱纯化即得到棕黄色粉末状固体,即为甲磺酸仑伐替尼杂质,化学名称为4-(4-氨基-3-氯苯氧基)-7-甲氧基-n,n-二甲基喹啉-6-羧酸,命名为m1z15,其结构式为
[0099]
经计算,产物质量为0.12g,产率为87%。
[0100]
通过实施例1-3可知,采用本发明提供的甲磺酸仑伐替尼杂质的制备方法制得的甲磺酸仑伐替尼杂质,合成产率高,所生成的产物纯度高,结构稳定,制备过程工艺简单、原料廉价且重现性好,十分利于工业扩大生产。
[0101]
本发明所提供的甲磺酸仑伐替尼杂质主要用于医药领域对甲磺酸仑伐替尼的临床、药理、药代动力学、毒理等方面的分析研究,对于仑伐替尼药品的质量控制具有重要意
义;同时所合成的两种甲磺酸仑伐替尼杂质可作为医药领域甲磺酸仑伐替尼类杂质或药物的中间体,进行其他仑伐替尼的合成制备,所述的合成方法亦可使用借鉴。
[0102]
请注意,以上实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上实施例仅表达了本技术的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干变形和改进,这些都属于本技术的保护范围。因此,本技术专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
