1.本发明属于制药技术领域,具体涉及一种阿米卡星杂质及其制备方法和应用。
背景技术:
2.阿米卡星,又名丁胺卡那霉素,分子量为585,其化学式为:
[0003][0004]
阿米卡星是一种半合成的氨基糖苷类抗生素,由卡那霉素结构修饰而得,硫酸阿米卡星为其硫酸盐。硫酸阿米卡星是我国基本药物目录收载的品种之一,是感染性疾病的常规用药。
[0005]
硫酸阿米卡星的最大日服用剂量为1.5g,根据ichq3b指导原则,硫酸阿米卡星注射液中未知杂质的鉴定限度为0.2%,硫酸阿米卡星注射液中若有含量超过0.2%的未知杂质,对硫酸阿米卡星注射液的质量会引入较大的风险,存在一定的安全性隐患,需对其结构进行鉴定,从而有效的控制产品质量。
[0006]
目前,现有技术中没有关于阿米卡星中杂质结构的相关报道,无法对硫酸阿米卡星注射液中超鉴定限的未知杂质进行定性定量研究,有效控制产品质量。
技术实现要素:
[0007]
本发明所要解决的技术问题是克服现有技术中的不足,提供一种阿米卡星杂质及其制备方法和应用,该阿米卡星杂质可以作为阿米卡星及其衍生物的杂质对照品,为阿米卡星及其衍生物的质量研究提供基础,尤其为硫酸阿米卡星注射液的质量研究提供基础。
[0008]
为解决以上技术问题,本发明采取的技术方案是:
[0009]
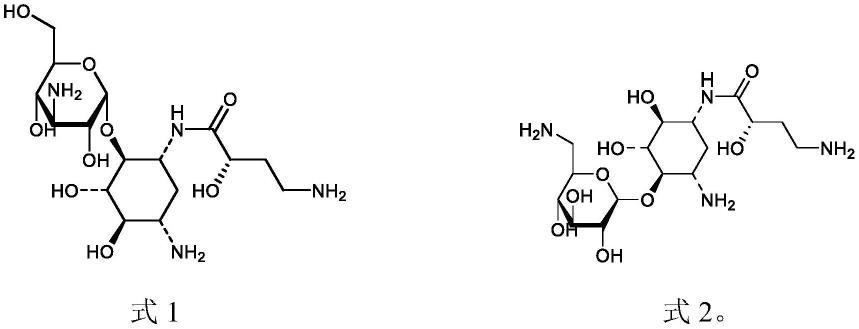
一种阿米卡星杂质,包括式1化合物,式2化合物,
[0010][0011]
为解决以上技术问题,本发明采取的又一技术方案是:
[0012]
一种阿米卡星杂质的制备方法,包括如下步骤:
[0013]
(1)使阿米卡星及其衍生物在酸热条件下制备杂质溶液;
[0014]
(2)对步骤(1)得到的杂质溶液进行分离,得到阿米卡星杂质制备液。
[0015]
优选地,阿米卡星衍生物包括硫酸阿米卡星、阿米卡星其他盐形式及制剂形式。
[0016]
优选地,在步骤(1)中,阿米卡星及其衍生物与酸溶液在105℃~120℃条件下进行化学反应,制备杂质溶液,反应时间为1~4.5小时;
[0017]
优选地,反应温度为105℃,反应时间为2小时。
[0018]
优选地,酸选自盐酸、硫酸和硝酸中的一种或多种;
[0019]
优选地,酸为盐酸;
[0020]
优选地,酸溶液的浓度为1mol/l。
[0021]
优选地,阿米卡星及其衍生物与酸溶液的质量体积比为1g:(1~5)ml;
[0022]
优选地,阿米卡星及其衍生物与酸溶液的质量体积比为1g:3ml。
[0023]
优选地,在步骤(2)中,对步骤(1)得到的杂质溶液进行高效液相色谱分离,色谱柱以十八烷基硅烷键合硅胶为填充剂,流动相为离子对试剂与乙腈的混合溶液;
[0024]
优选地,离子对试剂选用挥发性的离子对试剂,
[0025]
优选地,离子对试剂选用三氟乙酸、五氟丙酸和七氟丁酸中的一种;
[0026]
更优选地,离子对试剂选用五氟丙酸;
[0027]
优选地,离子对试剂与乙腈的体积比为90~70:10~30;
[0028]
更优选地,离子对试剂与乙腈的体积比为85:15。
[0029]
优选地,离子对试剂的浓度为0.1%~0.5%,优选为0.5%,离子对试剂的浓度为质量浓度m/v,单位优选为mg/ml。
[0030]
优选地,色谱柱的柱温为20~40℃,进样体积为10~100μl;流动相的流速为1~5ml/min;检测波长为200~220nm;目标杂质峰的接取时间为4.3~5.2min。
[0031]
优选地,制备方法还包括对步骤(2)得到的阿米卡星杂质制备液进行冷冻干燥,得到阿米卡星杂质固体。
[0032]
为解决以上技术问题,本发明采取的又一技术方案是:
[0033]
如上所述阿米卡星杂质作为阿米卡星及其衍生物的杂质对照品的应用,
[0034]
优选地,阿米卡星衍生物包括硫酸阿米卡星、阿米卡星其他盐形式及制剂形式。
[0035]
由于以上技术方案的采用,本发明与现有技术相比具有如下优点:
[0036]
1、本发明的技术方案通过对阿米卡星酸热破坏后进行液相制备,避免了合成过程中复杂的工艺操作及大量试剂的使用导致的环境污染;
[0037]
2、本发明提供一种阿米卡星杂质的制备方法,制备得到的阿米卡星杂质可以作为阿米卡星及其衍生物的杂质对照品,为阿米卡星及其衍生物的质量研究提供基础。
附图说明
[0038]
图1为本技术的目标杂质的峰形及保留时间的高效液相色谱特征标示图。
具体实施方式
[0039]
为使本发明的技术方案和有益效果能够更加明显易懂,下面通过列举具体实施例的方式进行详细说明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0040]
下列实施例中未注明具体实验步骤或条件者,按照本领域内的常规实验步骤的操作或条件即可进行。本发明所用物料、样品、耗材及设备,均为可以通过市购获得的常规产品,包括但不限于本技术实施例中采用的物料、样品、耗材及设备。
[0041]
本发明阿米卡星杂质,包括式1化合物,式2化合物,
[0042][0043]
本发明阿米卡星杂质的制备方法,包括如下步骤:
[0044]
(1)使阿米卡星及其衍生物在酸热条件下制备杂质溶液;
[0045]
(2)对步骤(1)得到的杂质溶液进行分离,得到阿米卡星杂质制备液,阿米卡星杂质包括式1化合物,式2化合物,
[0046]
[0047]
在某些实施例中,阿米卡星衍生物包括硫酸阿米卡星、阿米卡星其他盐形式及制剂形式。
[0048]
在某一具体的实施例中,使硫酸阿米卡星在酸热条件下制备杂质溶液。
[0049]
在某些实施例中,在步骤(1)中,硫酸阿米卡星与酸溶液在105℃~120℃条件下进行化学反应,反应时间为1~4.5小时,制备杂质溶液。
[0050]
在某一具体的实施例中,反应温度可以为105℃~115℃,105℃~110℃,更具体地,可以为105℃,110℃,115℃,120℃。
[0051]
在某一具体的实施例中,反应时间可以为1~4小时,1~3小时,1.5~3小时,1.5~2.5小时,更具体地,可以为1小时,1.5小时,2小时,3小时,4小时。
[0052]
在某些实施例中,步骤(1)中的酸选自盐酸、硫酸和硝酸中的一种或多种。
[0053]
在某一具体的实施例中,步骤(1)中的酸为盐酸。
[0054]
在某些实施例中,步骤(1)中酸溶液的浓度为1mol/l。
[0055]
在某一具体的实施例中,步骤(1)中盐酸的浓度为1mol/l。
[0056]
在某些实施例中,步骤(1)中硫酸阿米卡星与酸溶液的质量体积比为1g:(1~5)ml。
[0057]
在某一具体的实施例中,步骤(1)中硫酸阿米卡星与酸溶液的质量体积比为1g:3ml。
[0058]
在某一具体的实施例中,步骤(1)中硫酸阿米卡星与盐酸溶液的质量体积比为1g:3ml。
[0059]
在某些实施例中,在步骤(2)中,对步骤(1)得到的杂质溶液进行高效液相色谱分离,色谱柱以十八烷基硅烷键合硅胶为填充剂,流动相为离子对试剂与乙腈的混合溶液。
[0060]
在某些实施例中,离子对试剂选用挥发性的离子对试剂。
[0061]
在某些实施例中,离子对试剂选用三氟乙酸、五氟丙酸和七氟丁酸中的一种。
[0062]
在某一具体的实施例中,离子对试剂选用五氟丙酸。
[0063]
在某些实施例中,离子对试剂与乙腈的体积比为90~70:10~30。
[0064]
在某一具体的实施例中,离子对试剂与乙腈的体积比为85:15。
[0065]
在某些实施例中,五氟丙酸与乙腈的体积比为90~70:10~30。
[0066]
在某一具体的实施例中,五氟丙酸与乙腈的体积比为85:15。
[0067]
在某些实施例中,离子对试剂的浓度为0.1%~0.5%,离子对试剂的浓度为质量浓度m/v,单位优选为mg/ml,0.5%即5mg/ml。
[0068]
在某一具体的实施例中,离子对试剂的浓度为0.5%。
[0069]
在某些实施例中,五氟丙酸的浓度为0.1%~0.5%。
[0070]
在某一具体的实施例中,五氟丙酸的浓度为0.5%,五氟丙酸的浓度为体积浓度v/v,0.5%即每1l溶液中含有5ml五氟丙酸。
[0071]
在某些实施例中,色谱柱为zorbax sb-c18,柱温为20~40℃,可以为30~40℃。
[0072]
在某些实施例中,进样体积为10~100μl,可以为30~100μl,50~100μl。
[0073]
在某些实施例中,流动相的流速为1~5ml/min,可以为1~4ml/min。
[0074]
在某些实施例中,检测波长为200~220nm,可以为200~217nm。
[0075]
在某些实施例中,目标杂质峰的接取时间为4.3~5.2min。
[0076]
在某一具体的实施例中,色谱柱为zorbax sb-c18(9.4mm
×
25cm,5μm);柱温为30℃;进样体积为50μl;流动相的流速为4ml/min;检测波长为217nm;目标杂质峰的接取时间为4.3~5.2min。
[0077]
在某些实施例中,制备方法还包括对步骤(2)得到的阿米卡星杂质制备液进行冷冻干燥,得到阿米卡星杂质固体。
[0078]
如上所述的阿米卡星杂质作为阿米卡星及其衍生物的杂质对照品的应用。
[0079]
在某一具体的实施例中,阿米卡星衍生物包括硫酸阿米卡星、阿米卡星其他盐形式及制剂形式。
[0080]
本发明中涉及的物料、设备、样品信息如表1至表3所示,其中原料药和药用辅料均符合《中国药典》2020年版的相关规定。
[0081]
表1物料信息
[0082]
名称来源批号超纯水milli-q direct8自制/磷酸二氢钾上海凌峰化学试剂有限公司20200309乙腈tedia21075179磷酸国药集团化学试剂有限公司20210107辛烷磺酸钠永华化学股份有限公司20210312无水硫酸钠西陇科学股份有限公司2105122
[0083]
表2设备信息
[0084][0085]
表3样品信息
[0086][0087]
实验例1反应温度选择
[0088]
称取原料(硫酸阿米卡星)1g,置于具塞试管中,分别加入1mol/l盐酸溶液5ml,置
于105℃、120℃烘箱,分别在1小时、2小时取出,放冷置室温,加水稀释至刻度10ml,摇匀,照下述色谱条件进样检测:
[0089]
用十八烷基硅烷键合硅胶为填充剂(spursil柱,4.6mm
×
250mm,5μm);取辛烷磺酸钠1.8g和无水硫酸钠20.0g,加ph为3.0的0.2mol/l磷酸盐缓冲液(0.2mol/l磷酸二氢钾溶液,用0.2mol/l磷酸溶液调节ph值至3.0)50ml和水900ml溶解,加乙腈50ml,混匀,作为流动相a;取辛烷磺酸钠1.8g和无水硫酸钠20.0g,加ph为3.0的0.2mol/l磷酸盐缓冲液50ml和水850ml溶解,加乙腈92ml,混匀,作为流动相b,流动相a-流动相b(50:50);流动相流速为1.3ml/min;柱温为40℃;检测波长为200nm,进样体积为10μl。结果如表4所示:
[0090]
表4不同反应温度实验结果(峰面积)
[0091][0092]
根据实验结果可知,120℃条件下,目标未知杂质及阿米卡星降解速度偏快,不利于后期杂质分离,故选择反应温度为105℃。
[0093]
实验例2盐酸加入量的选择
[0094]
称取原料(硫酸阿米卡星)1g,置于具塞试管中,分别加入1mol/l盐酸1ml、3ml、5ml,置于105℃烘箱,分别在1小时、1.5小时、2小时、2.5小时(加入量为1ml的条件下,额外考察了3小时的反应时间点)取出,放冷置室温,加水稀释至刻度10ml,摇匀,参照实验例1的色谱条件进样检测,结果如表5所示:
[0095]
表5不同盐酸溶液加入量实验结果(峰面积)
[0096][0097]
根据实验结果可知,1mol/l盐酸溶液加入量为1ml时,反应时间过长,加入量为3ml及5ml时,反应所需时间基本相同,故选择1mol/l盐酸加入量为3ml及5ml。
[0098]
实验例3反应时间的选择
[0099]
称取原料(硫酸阿米卡星)1g,置于具塞试管中,加1mol/l盐酸3ml超声溶解,置于105℃烘箱,分别于各反应时间点(1小时、1.5小时、2小时、2.5小时、3.5小时、4小时、4.5小时)取样250μl,放冷置室温,取样20μl,加980μl水,混合均匀,照实验例1的色谱条件进样检测,结果如表6所示:
[0100]
表6不同反应时间实验结果(峰面积)
[0101][0102]
根据实验结果可知,随反应时间延长,在2.5小时后,目标未知杂质峰面积无明显变化,最优反应时间为1.5~2.5小时,但其后有一干扰杂质,随时间延长,呈增长趋势,不利于后期制备分离,故将反应时间定为2小时。
[0103]
综上所述,杂质溶液的制备方法为:称取硫酸阿米卡星原料1g,置于具塞试管中,加1mol/l盐酸溶液3ml溶解后,置于105℃放置2小时。
[0104]
实验例4液相制备方法开发
[0105]
发明人发现目标未知杂质及阿米卡星极性较强,在c18柱上保留较弱,采用常规流动相较难分离。为增强目标未知杂质及阿米卡星的保留,发明人发现在流动相中加入离子对试剂,可以增加目标未知杂质及阿米卡星的保留,同时考虑后期的杂质纯化,采用挥发性的离子对试剂。
[0106]
三氟乙酸为常用的挥发性离子对试剂,但其离子对作用较弱,多次试验(调整比例、柱温、增加缓冲盐)均未获得较好的结果,目标未知杂质与阿米卡星保留较弱,且无法有效分离。发明人采用离子对作用更强的五氟丙酸和七氟丁酸进行试验。
[0107]
色谱条件具体为:用十八烷基硅烷键合硅胶为填充剂(spursil柱,4.6mm
×
250mm,5μm);以0.1%五氟丙酸及0.1%七氟丁酸溶液分别作流动相a,乙腈作流动相b,流速为1.0ml/min,检测波长为200nm,柱温为30℃,进样体积为10μl。
[0108]
取硫酸阿米卡星原料1g,置于具塞试管中,加1mol/l盐酸溶液3ml溶解后,于105℃放置2小时后,取出,放冷至室温,作为杂质溶液。取上述溶液20μl置于进样瓶中,加980μl水,混匀,作为杂质实验溶液;取硫酸阿米卡星原料50mg,置于10ml量瓶中,加水溶解并稀释至刻度,摇匀,作为阿米卡星定位溶液。分别采用五氟丙酸及七氟丁酸在不同流动相比例条件下进行实验,结果如表7所示(其中,流动相a与流动相b的比例为体积比):
[0109]
表7不同流动相实验结果
[0110][0111]
根据实验结果可知,0.1%五氟丙酸-乙腈(90:10)条件下(条件
③
),目标未知杂质与阿米卡星保留时间适宜,分离度较好,所以选择五氟丙酸作为流动相做进一步优化。
[0112]
由于0.1%五氟丙酸-乙腈(90:10)条件下运行时间偏长,为提高制备效率,对流动相比例做进一步优化。取杂质溶液,在0.1%五氟丙酸-乙腈(85:15)条件下进样,结果流动相比例为85:15时,目标未知杂质与阿米卡星峰形较好,两化合物可有效分离,保留时间适宜,可以缩短运行时间,提高制备效率,故选择流动相比例为0.1%五氟丙酸-乙腈的体积比为85:15。
[0113]
为提高进样量,获得更大的制备效率,可以更换更大内径的色谱柱(zorbax sb-c18,9.4mm
×
25cm,5μm),由于色谱柱内径变大,可以将流速对应调整为4.0ml/min。
[0114]
为提高制备效率,提高进样浓度。取杂质溶液200μl,置于进样瓶中,加800μl水,混匀,作为杂质实验溶液,照上述色谱条件进样。
[0115]
提高进样浓度后,目标未知杂质和阿米卡星峰均分叉,应为离子对试剂负载能力不够导致(样品量远大于离子对试剂的结合能力,导致部分样品保留变弱,出峰提前,表现出峰形分叉现象),提高流动相中五氟丙酸浓度至0.5%后重新进样,结果无响应,查看紫外吸收谱图发现,在0.5%五氟丙酸条件下,最大吸收往长波长偏移,最大吸收波长为217nm。
[0116]
根据试验结果可知,提高五氟丙酸浓度为0.5%后,流动相负载能力增加,目标未知杂质和阿米卡星峰形和分离度均变好,在选择五氟丙酸浓度为0.5%时,需同时将检测波长调整为217nm。
[0117]
实验例5进样体积及制备液接取时间的选择
[0118]
称取硫酸阿米卡星原料1g,置于具塞试管中,加1mol/l盐酸溶液3ml溶解后,于105℃放置2小时后,取出,放冷至室温,量取200μl置于进样瓶中,加800μl水,混匀,照实验例1的色谱条件进样检测,进样体积分别为10μl、30μl、50μl、100μl。
[0119]
随着进样体积增加,目标未知杂质及阿米卡星峰保留时间不断提前,应为样品量
增加导致离子对试剂负载能力变弱导致,阿米卡星峰保留时间提前可能干扰目标未知杂质的制备,为进一步确认进样体积及接取时间,分别进样30μl、50μl、100μl,在检测器出口端手动接取洗脱液,接取时间段如表8所示:
[0120]
表8不同进样体积接取杂质时间段
[0121]
进样体积时间段
①
时间段
②
时间段
③
时间段
④
时间段
⑤
30μl4.2~4.54.5~4.84.8~5.15.1~5.45.4~5.750μl4.3~4.64.6~4.94.9~5.25.3~5.65.6~5.9100μl4.2~4.54.6~4.94.9~5.25.3~5.65.6~5.9
[0122]
取接取的洗脱液,在实验例1的色谱条件进样检测,记录各时间段目标未知杂质及阿米卡星的峰面积,结果如表9所示:
[0123]
表9不同接取时间段检测结果(峰面积)
[0124][0125]
根据实验结果可知,当进样体积为30μl时,目标未知杂质集中的时间段为4.2~5.4min,总峰面积为92.8,阿米卡星对目标未知杂质制备基本无干扰;当进样体积为50μl时,目标未知杂质集中的时间段为4.3~5.2min,总峰面积为128.3,阿米卡星对目标未知杂质制备干扰较小;当进样体积为100μl时,目标未知杂质集中的时间段为4.2~5.6min,总峰面积为133.7,阿米卡星对目标未知杂质制备有较大干扰。综合目标未知杂质的制备效率及阿米卡星的干扰程度,将进样体积定为50μl,接取时间定为4.3~5.2min。
[0126]
实验例6目标杂质定性研究
[0127]
取接取的洗脱液进行冷冻干燥,得到白色疏松块状固体,取该杂质进行质谱及核磁定性研究。
[0128]
采用二级质谱(ms/ms)对目标杂进行定性解析,得到其分子量约为424,425.2为其[m h]
,447.2为其[m na]
。根据二级质谱碎片信息,对其结构进行推测。推测其结构为式1或式2所示化合物。
[0129][0130]
二级质谱碎片对应的信息如表10所示:
[0131]
表10目标杂质二级质谱信息
[0132][0133][0134]
取目标杂质对照品,进行重水交换核磁共振氢谱试验,结果如表11所示:
[0135]
表11目标杂质核磁共振氢谱信息
[0136][0137]
根据核磁共振氢谱实验结果,5.51及5.15位移分别归属羰基异头碳上的o-ch-o,基本可以判定,目标杂质是式1、式2化合物组成的混合物。
[0138]
实施例1
[0139]
称取硫酸阿米卡星原料1g,置于具塞试管中,加1mol/l盐酸溶液3ml溶解后,于105℃放置2小时,取出,放冷至室温,取200μl,置于进样瓶中,加800μl水,混匀,作为杂质溶液。用十八烷基硅烷键合硅胶为填充剂(zorbax sb-c189.4mm
×
25cm,5μm);以0.5%五氟丙酸-乙腈(体积比为85:15)作流动相;流速为4.0ml/min;柱温为30℃;检测波长为217nm,精密量取杂质溶液50μl,注入色谱仪,在检测器出口端手动接取4.3~5.2min的洗脱液。对洗脱液进行冷冻干燥,得到白色疏松块状固体,即为包括式1化合物,式2化合物的阿米卡星杂质。
[0140]
应当理解,以上实施例均为示例性的,不用于包含权利要求所包含的所有可能的实施方式。在不脱离本公开的范围的情况下,还可以在以上实施例的基础上做出各种变形和改变。同样的,也可以对以上实施例的各个技术特征进行任意组合,以形成可能没有被明确描述的本发明的另外的实施例。因此,上述实施例仅表达了本发明的几种实施方式,不对本发明专利的保护范围进行限制。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。