mtor抑制剂、制备方法及用途
技术领域
1.本发明涉及mtor抑制剂、制备方法及用途,属于化学药物技术领域。
背景技术:
2.哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mtor),属于丝氨酸/苏氨酸蛋白激酶,在生物细胞中以mtorc1(mtor complex 1)和mtorc2(mtor complex 2)两种复合物形式广泛存在。mtor在细胞的生长、凋亡、自噬中都扮演重要角色,作为细胞内多个信号通路的重要组成部分,参与调控机体生长和代谢平衡,故mtor抑制剂被广泛用于广谱抗菌、抗病毒、免疫抑制和抗肿瘤等领域。
3.第一代mtor抑制剂,例如雷帕霉素及其衍生似物“rapalogs”,是以构象改变的方式阻止mtor激活的mtorc1抑制剂;例如依维莫司(everolimus),可与胞内蛋白fkbp12结合形成抑制性的复合体mtorc1,可抑制mtor的活性,mtor信号通路的抑制可导致转录调节因子s6核糖体蛋白激酶(s6k1)和真核生物延伸因子4e-结合蛋白(4e-bp1)的活性降低,从而干扰细胞周期、血管新生、糖酵解等相关蛋白的翻译和合成。但由于此类化合物在抑制mtorc1的同时,激活了akt反馈调节通路,限制了其在临床上的广泛应用,且产生了耐药性。因此,为了更好的满足临床与市场的需求,开发出一种新的更有效、抑制活性更好的mtor抑制剂具有重要的经济效益和社会效益。
技术实现要素:
4.针对现有技术的上述不足,本发明的目的在于提供一种mtor抑制剂、制备方法及用途。本发明涉及的mtor抑制剂更有效、抑制活性更好,用于预防和/或治疗由mtor信号通路功能失调而导致的恶性肿瘤、免疫性疾病、病毒感染、神经性疾病等,副作用较小。具体技术方案如下:第一方面,具有式ι所示结构的化合物或其药学上可接受的盐:
式中,r1选自、、、;r2选自c
1-c6烷基、环丙基、环丁基、环戊基、环己基、、、、、、;r3选自h、oh、och3、och2ch3、och2ch2ch3、och(ch3)2、、。
5.第二方面,式ι所示结构的化合物或其药学上可接受的盐的制备方法,包括如下步骤:
式中,r1、r2、r3的定义同第一方面对r1、r2、r3的定义。
6.步骤1)、化合物vii的合成化合物viii、还原剂于反应溶剂中进行还原反应,得到化合物vii;进一步地,在步骤1)中,所述反应温度为20~30℃,例如,反应温度可以为20℃、25℃或30℃等;当反应温度过低时,反应速率较慢,反应时间较长;当反应温度过高时,会影响化合物vii(如式vii所示的化合物)的稳定性。所述还原剂为铁粉、锌粉、钯碳、ni、氢气中的至少一种,优选是钯碳、氢气的组合。所述反应溶剂为乙醇、甲醇、二氧六环、四氢呋喃中的至少一种,优选是甲醇。
7.通过控制反应温度、选择特定还原剂等手段,最终化合物vii的收率能达61.2%。
8.步骤2)、化合物v的合成化合物vii、化合物vi于反应溶剂中进行反应,得到化合物v;进一步地,在步骤2)中,所述反应温度为0~25℃;在加料阶段的温度控制为0~5℃,其目的是降低反应物活性,抑制副反应;如果主反应的反应温度过低,也导致反应速率较慢,反应时间较长;因此,需要对主反应升温,但如果反应温度过高,副反应得不到抑制,也会影响最终的收率;最终主反应的最优反应温度控制在25℃。所述反应溶剂为n,n-二甲基甲酰胺、四氢呋喃中的至少一种,优选四氢呋喃。
9.通过控制反应温度,最终化合物v的收率能达81.1%。
10.步骤3)、化合物iv的合成化合物v、还原剂于反应溶剂中进行还原反应,得到化合物iv;进一步地,在步骤3)中,所述反应温度为-30℃~0℃;在加料阶段的温度控制为-30~-20℃,其目的是降低反应物活性,抑制副反应;但是,如果主反应的反应温度过低,会导致反应速率较慢,反应时间较长;因此,需要对主反应升温,但如果反应温度过高,副反应会得
不到抑制,会影响最终的收率;最终主反应的最优反应温度控制在-10℃。所述还原剂为( )-二异松蒎基氯硼烷、硼氢化钠、三乙酰基硼氢化钠中的至少一种,还原剂优选为( )-二异松蒎基氯硼烷。所述反应溶剂为n,n-二甲基甲酰胺、四氢呋喃、甲苯、n-甲基吡咯烷酮中的至少一种,反应溶剂优选为四氢呋喃。
11.通过控制反应温度、选择特定还原剂等手段,最终化合物iv的收率能达71.1%。
12.步骤4)、化合物iii的合成化合物iv、酸于反应溶剂中进行水解反应,得到化合物iii;进一步地,在步骤4)中,所述反应温度为0℃~60℃,例如,反应温度可以为0℃、10℃、20℃、30℃、40℃、50℃或60℃等;所述酸为盐酸、三氟乙酸中的至少一种,优选为三氟乙酸。所述反应溶剂为二氯甲烷、甲醇、乙醇中的至少一种,优选为二氯甲烷。采用三氟乙酸脱氨基上的boc(叔丁氧羰基的缩写)效率最高;另外,要使外消旋化程度减到最小,需要使用较低的反应温度,例如最优反应温度控制在0℃。
13.通过控制反应温度、选择特定酸等手段,最终化合物iii的收率能达80.1%。
14.步骤5)、化合物ii-1的合成化合物ii-3、化合物ii-2、碱于反应溶剂中在40~50℃的温度下进行偶联反应,反应完毕之后,再加入酸后在10~40℃继续反应(在酸性条件下进行脱保护基关环反应),得到化合物ii-1;进一步地,在步骤5)中,所述碱为三乙胺、二异丙基乙胺中的至少一种,优选为三乙胺;所述酸为盐酸、三氟乙酸中的至少一种,优选为盐酸;所述反应溶剂为二氯甲烷、n,n-二甲基甲酰胺中的至少一种。在偶联反应中,采用三乙胺的反应效率最高,反应温度可以为40℃、43℃、46℃或50℃等,优选为43
±
3℃。采用盐酸进行关环反应的效率最高;另外,由于还涉及脱保护基反应,因此需要降温,例如最优反应温度控制在20
±
5℃。
15.通过控制偶联反应温度、脱保护基关环反应温度、选择特定酸、碱等手段,化合物ii-1的收率能达84.0%。
16.步骤6)、化合物ii的合成化合物ii-1、碱于反应溶剂中进行水解反应,得到化合物ii;进一步地,在步骤6)中,所述反应温度为20~80℃,例如,反应温度可以为20℃、25℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃或80℃等,优选为25℃;所述碱为氢氧化钠、氢氧化钾、氢氧化锂、碳酸钾、叔丁醇钾、甲醇钠中的至少一种,优选为氢氧化钠;所述反应溶剂为四氢呋喃、甲醇、乙醇、二氧六环中的至少一种,优选为乙醇。
17.通过控制反应温度、选择特定碱等手段,化合物ii的收率能达79.3%。
18.步骤7)、化合物i-1的合成化合物ii、化合物iii、缚酸剂、缩合剂于反应溶剂中进行反应,得到化合物i-1;进一步地,在步骤7)中,所述反应温度为0℃~60℃;在将化合物ii、化合物iii、缚酸剂添加到反应溶剂中时,通过降温至0℃来抑制副反应;但如果主反应的反应温度过低,会导致反应速率较慢,反应时间较长;因此,需要对主反应升温,但如果反应温度过高,副反应会得不到抑制,会影响最终的收率;因此,在加入缩合剂后,主反应温度需升温至25℃。缚酸剂为4-二甲氨基吡啶、n,n-二异丙基乙胺中的至少一种,优选4-二甲氨基吡啶。所述缩合剂为2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、o-苯并三氮唑-四甲基脲六
氟磷酸酯、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、n,n'-二环己基碳酰亚胺中的至少一种,优选n,n'-二环己基碳酰亚胺。所述反应溶剂为n,n-二甲基甲酰胺、二氯甲烷、四氢呋喃中的至少一种,优选二氯甲烷。
19.通过控制反应温度、选择特定缚酸剂、缩合剂等手段,化合物i-1的收率能达55.6%。
20.进一步地,当r3选自时:化合物i-1、乙酸酐(c4h6o3)于反应溶剂中进行反应,得到化合物i-2,化合物i-2与化合物i是同种化合物;其中,所述反应温度为20~30℃,反应温度不宜过高,否则将会增加副产物,因此,反应温度优选为20℃;所述反应溶剂为n,n-二甲基甲酰胺、二氯甲烷、四氢呋喃、甲苯中的至少一种,优选甲苯。
21.通过控制反应温度,选择乙酸酐作为酰化剂等手段,化合物i-2的收率能达65.0%。
22.进一步地,当r3选自时:化合物i-1、氯甲酸苄酯(cbz-cl)、碱于反应溶剂中进行反应,得到化合物i-3,化
合物i-3与化合物i是同种化合物;其中,所述反应温度为15~30℃,碱选自碳酸铯、碳酸钾、碳酸氢钠中的至少一种,优选为碳酸氢钠;所述反应溶剂为n,n-二甲基甲酰胺、二氯甲烷、四氢呋喃中的至少一种,优选为四氢呋喃。
23.通过控制反应温度、选择特定碱等手段,化合物i-3的收率能达78.3%。
24.进一步地,当r3选自h时,化合物i-1与化合物i是同种化合物。
25.第三方面,一种mtor抑制剂,包括:具有式ι所示结构的化合物或其药学上可接受的盐,以及一种或多种药学上可接受的载体或稀释剂。
26.第四方面,具有式ι所示结构的化合物或其药学上可接受的盐在制备预防和/或治疗恶性肿瘤、免疫性疾病、病毒感染、神经性疾病药物中的用途。其中,所述恶性肿瘤为恶性胶质瘤、肾恶性肿瘤、胃恶性肿瘤、恶性皮肤肿瘤、横纹肌肉瘤、膀胱恶性肿瘤、乳腺恶性肿瘤、子宫恶性肿瘤、肺恶性肿瘤、结肠恶性肿瘤、前列腺恶性肿瘤、卵巢恶性肿瘤或胰腺恶性肿瘤。
27.本发明的有益效果:1)、式i所示的化合物、药学上可接受的盐以及含有这些化合物作为活性成分的药物组合物作为mtor抑制剂,更有效、抑制活性更好,用于预防和/或治疗由mtor信号通路功能失调而导致的恶性肿瘤、免疫性疾病、病毒感染、神经性疾病等,副作用较小;可以通过向需要此种预防和/或治疗的患者给予治疗有效剂量的一种或者多种式i所示的化合物,达到治疗的目的。
28.2)、本发明的制备方法简单,条件温和,操作方便,设备条件要求不高,极易实现,且后处理简单,副产物少,收率高,适用于工业化大规模生产,具有极大的应用价值。
具体实施方式
29.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
30.定义“药学上可接受的盐”是指那些保留母体化合物的生物有效性及特性的盐。该盐包括:酸加成盐,其是通过母体化合物的游离碱与无机酸或与有机酸的反应而获得的;所述无机酸诸如盐酸、氢溴酸、氢碘酸、硝酸、磷酸、硫酸及高氯酸等;所述有机酸诸如乙酸、草酸、(d)或(l)苹果酸、马来酸、甲烷磺酸、乙烷磺酸、对甲苯磺酸、水杨酸、酒石酸、苯磺酸(苯磺酸盐)、苯甲酸、樟脑磺酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、羟乙磺酸、乳酸、马来酸、扁桃酸、黏液酸、双羟萘酸、泛酸、琥珀酸、酒石酸或丙二酸等;优选为盐酸或(l)-苹果酸;或者当母体化合物中存在的酸质子被置换为金属离子或与有机碱配位时,形成盐,所述金属离子例如碱金属离子、碱土金属离子或铝离子;所述有机碱诸如乙醇胺、二乙醇胺、三乙醇胺、缓血酸胺、n-甲基葡糖胺及类似物。
[0031]“药学上可接受的载体”是指药学领域常规的药物载体,对生物不产生明显刺激且不会消除所给予的化合物的生物活性及特性的载体,例如:稀释剂,如水等;填充剂,如淀粉、蔗糖等;粘合剂,如纤维素衍生物、藻酸盐、明胶、聚乙烯吡咯烷酮;湿润剂,如甘油;崩解
剂,如琼脂、碳酸钙和碳酸氢钠;吸收促进剂,如季铵化合物;表面活性剂,如十六烷醇;吸附载体,如高岭土和皂粘土;润滑剂,如滑石粉、硬脂酸钙和硬脂酸镁和聚乙二醇等。另外,还可以在上述药物组合物中加入其它辅料,如香味剂和甜味剂等。
[0032]
本发明的化合物可具有一个或多个不对称中心;该化合物因此可以个别(r)-立体异构体或(s)-立体异构体形式制备或以其混合物形式制备。除非另有说明,否则本说明书及权利要求中的特定化合物的描述或名称意欲包括个别对映异构体与其外消旋混合物或其它混合物。用于测定立体化学构型及分离立体异构体的方法在本领域中是熟知的(参见"advanced organic chemistry"的第4章中的论述,第4版,j. march,john wiley及sons,new york,1992)。因此,本发明亦涵盖具有mtor抑制活性的任何立体异构形式、其相应对映异构体(d-异构体及l-异构体或( )异构体及(-)异构体)及其非对映异构体及其混合物且不限于任一种立体异构形式。
[0033]“室温”具有本领域公知的含义,一般是指24~28℃。
[0034]
本发明的实施例提供了一种具有式ⅰ结构的化合物或其药学上可接受的盐:其中,当r3选自h时,r1选自、或,r2选自、、或;当r3选自或时,r1选自,r2选自。
[0035]
进一步地,式ⅰ所示结构可以为但不限于下述结构:
。
[0036]
本发明先后进行过多次试验,现列举其中一部分试验结果作为参考对发明进行详细描述,下面结合具体实施例进行详细说明。
[0037]
实施例1第一步:将化合物viii(31.3g,0.1mol)、钯碳(3.1g)加入甲醇(300ml)中搅拌,通入氢气,氢气的压力保持在40~50psi,搅拌完毕后升至25℃,保温反应12小时,tlc监测反应,反应完毕后,过滤得第一固体物,第一固体物加入到500ml甲醇中,加热至回流,热过滤得滤液,减压浓缩得第二固体物,第二固体物加入到100ml乙酸乙酯和100ml石油醚混合溶液中,加热回流,降温析晶,得到17.3g黄色固体状的化合物vii,收率61.2%。其中,化合物viii的结构式如式viii所示,其他化合物以此类推。
[0038]
第二步:将化合物vii(15.0g,0.05mol)、化合物vi(二碳酸二叔丁酯,24.1g,0.11mol)溶于四氢呋喃(400ml)中,降温至0~5℃,溶解完毕后升至25℃,保温反应6小时,tlc监测反应,反应完毕后加水(100ml)淬灭反应,分液,取有机相,有机相用无水硫酸钠干燥,过滤,减压浓缩得到15.5g油状物,纯化后得灰色固体化合物v,收率81.1%。
[0039]
第三步:将化合物v(14.2g,0.04mol)溶于四氢呋喃(400ml)中,降温至-30℃,滴加( )-二异松蒎基氯硼烷(19.2g,0.06mol),滴加过程保温在-20~-30℃;升温至-10℃,保温反应16小时,反应完毕后,减压浓缩,加入乙醚和二乙醇胺(77.6ml,0.8mol),在室温搅拌4小时,过滤得有机相,浓缩,经柱层析(洗脱剂:二氯甲烷/甲醇=20/1),得10.9g油状的化合物iv,收率为71.1%。
[0040]
第四步:将化合物iv(10.0g,0.03mol)溶于二氯甲烷(100ml)中,降温至0℃,滴加三氟乙酸(50ml),升至室温搅拌反应2小时,tlc监测反应,反应完毕后,减压浓缩除去二氯甲烷及三氟乙酸,调节ph至8~10,析出固体,过滤、干燥得到5.9g固体状的化合物iii,收率为80.1%。
[0041]
第五步:将化合物1-ii-3(7.0g,0.02mol)、化合物1-ii-2(5.1g,0.02mol)溶于二氯甲烷(100ml)中,于40~46℃下缓慢滴加三乙胺(1.0g,0.04mol),搅拌反应3小时,tlc监测反应,反应完毕后,降温至15~25℃,滴加32ml 6n盐酸水溶液,反应完全后静置分层,有机相用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,有机相干燥,浓缩,得到8.2g固体状的化合物1-ii-1,收率为84.0%。
[0042]
第六步:
将化合物1-ii-1(8.0g,0.02mol)溶于乙醇(100ml)中,室温下搅拌加入氢氧化钠水溶液(1.6g,0.04mol),tlc监测反应,反应完毕后减压浓缩,加入2n盐酸水溶液调节ph至1~2,用乙酸乙酯(100ml
×
2)萃取,合并有机层,无水硫酸钠干燥,浓缩,得到5.8g类白色固体状的化合物1-ii,收率为79.3%。
[0043]
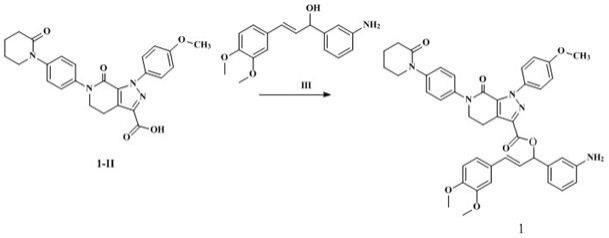
第七步:将化合物1-ii(3.6g,7.9mmol)溶于二氯甲烷(10ml),分别加入化合物iii(2.3g,7.9mmol)和dmap(4-二甲氨基吡啶,1.1g,8.7mmol)并降温至0℃,加入dcc(n,n'-二环己基碳酰亚胺,1.8g,8.7mmol),缓慢升至室温,继续反应2小时,tlc监测反应,反应完成后,加入乙酸乙酯,过滤得滤液,滤液减压浓缩得油状物,经柱层析得到3.2g类白色固体状的化合物1,收率为55.6%,esi( )m/z=728.8[m h]
。
[0044]
实施例2第一步:将化合物2-ii-3(7.4g,0.02mol)、化合物1-ii-2(5.1g,0.02mol)溶于二氯甲烷(100ml)中,于40~46℃下缓慢滴加三乙胺(1.0g,0.04mol),搅拌反应3小时,tlc监测反应,反应完毕后,降温至15~25℃,滴加32ml 6n盐酸水溶液,反应完全后静置分层,有机相用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,有机相干燥,浓缩,得到8.2g固体状的化合物
2-ii-1,收率为82.1%。
[0045]
第二步:将化合物2-ii-1(8.0g,15.6mmol)溶于乙醇(100ml)中,室温下搅拌加入氢氧化钠水溶液(1.3g,31.8mmol),tlc监测反应,反应完毕后减压浓缩,加入2n盐酸水溶液调节ph至1~2,用乙酸乙酯(100ml
×
2)萃取,合并有机层,无水硫酸钠干燥,浓缩,得到5.6g类白色固体状的化合物2-ii,收率为76.1%。
[0046]
第三步:将化合物2-ii(3.8g,8.0mmol)溶于二氯甲烷(20ml),分别加入化合物iii(2.4g,8.0mmol)和dmap(4-二甲氨基吡啶,1.2g,8.0mmol)并降温至0℃,加入dcc(n,n'-二环己基碳酰亚胺,1.8g,8.8mmol),缓慢升至室温,继续反应2小时,tlc监测反应,反应完成后,加入乙酸乙酯,过滤得到滤液,滤液减压浓缩得油状物,经柱层析得到2.7g类白色固体状的化合物2,收率为45.1%,esi( )m/z=742.8[m h]
。
[0047]
实施例3第一步:将化合物3-ii-3(0.7g,1.89mmol)、化合物1-ii-2(0.5g,1.89mmol)溶于二氯甲烷(10ml)中,于40~46℃下缓慢滴加三乙胺(0.4g,3.8mol),搅拌反应3小时,tlc监测反应,反
应完毕后,降温至15~25℃,滴加5ml 6n盐酸水溶液,反应完全后静置分层,有机相用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,有机相干燥,浓缩,得到0.8g固体状的化合物3-ii-1,收率为84%。
[0048]
第二步:将化合物3-ii-1(0.5g,1mmol)溶于乙醇(10ml)中,室温下搅拌加入氢氧化钠水溶液(0.12g,3mmol),tlc监测反应,反应完毕后减压浓缩,加入2n盐酸水溶液调节ph至1~2,用乙酸乙酯(50ml
×
2)萃取,合并有机层,无水硫酸钠干燥,浓缩,得到0.34g类白色固体状的化合物3-ii,收率为72.1%。
[0049]
第三步:将化合物3-ii(0.3g,0.6mmol)溶于二氯甲烷(5ml),分别加入化合物iii(0.17g,0.6mmol)和dmap(4-二甲氨基吡啶,0.1g,0.7mmol)并降温至0℃,加入dcc(n,n'-二环己基碳酰亚胺,0.14g,0.7mmol),缓慢升至室温,继续反应2小时,tlc监测反应,反应完成后,加入乙酸乙酯,过滤得滤液,滤液减压浓缩得油状物,经柱层析得到0.18g类白色固体状的化合物3,收率为41.1%,esi( )m/z=742.8[m h]
。
[0050]
实施例4第一步:
将化合物4-ii-3(1.0g,2.8mmol)、化合物1-ii-2(0.7g,2.8mmol)溶于二氯甲烷(5ml)中,于40~46℃下缓慢滴加三乙胺(0.6g,5.6mol),搅拌反应3小时,tlc监测反应,反应完毕后,降温至15~25℃,滴加4ml 6n盐酸水溶液,反应完全后静置分层,有机相用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,有机相干燥,浓缩,得到1.1g固体状的化合物4-ii-1,收率为80%。
[0051]
第二步:将化合物4-ii-1(0.7g,1.4mmol)溶于乙醇(5ml)中,室温下搅拌加入氢氧化钠水溶液(0.17g,4.2mmol),tlc监测反应,反应完毕后减压浓缩,加入2n盐酸水溶液调节ph至1~2,用乙酸乙酯(50ml
×
2)萃取,合并有机层,无水硫酸钠干燥,浓缩,得到0.45g类白色固体状的化合物4-ii,收率为70%。
[0052]
第三步:将化合物4-ii(0.4g,0.9mmol)溶于二氯甲烷(5ml),分别加入化合物iii(0.26g,0.9mmol)和dmap(4-二甲氨基吡啶,0.12g,1mmol)并降温至0℃,加入dcc(n,n'-二环己基碳酰亚胺,0.2g,1mmol),缓慢升至室温,继续反应2小时,tlc监测反应,反应完成后,加入乙酸乙酯,过滤得滤液,滤液减压浓缩所得油状物,经柱层析得到0.3g固体状的化合物4,收率为44.1%,esi( )m/z=730.4[m h]
。
[0053]
实施例5
将化合物1(0.3g,0.4mmol)溶于甲苯(4ml),升温至50℃搅拌溶清,然后降温至20℃,滴加乙酸酐(c4h6o3,81.6mg,0.8mmol),滴加结束后保温继续反应,tlc监测反应,反应完成后,加入碳酸氢钠水溶液,搅拌30分钟,加入乙酸乙酯,搅拌分层,有机相减压浓缩,得到0.2g油状的化合物5,收率为65.0%,esi( )m/z=770.8[m h]
。
[0054]
实施例6将化合物1(0.3g,0.4mmol)溶于四氢呋喃(4ml),加入碳酸氢钠(67.2mg,0.8mmol)、cbz-cl(氯甲酸苄酯,107.4mg,0.6mmol),在室温下继续反应,tlc监测反应,反应完成后,加入水搅拌30分钟,加入乙酸乙酯,搅拌分层,有机相用无水硫酸钠干燥,减压浓缩,得到0.27g油状的化合物6,收率为78.3%,esi( )m/z=862.9[m h]
。
[0055]
实施例7第一步:将化合物7-ii-3(1.0g,2.8mmol)、化合物7-ii-2(730.8mg,2.8mmol)溶于二氯甲烷(5ml)中,于40~46℃下缓慢滴加三乙胺(0.6g,5.6mol),搅拌反应3小时,tlc监测反应,反应完毕后,降温至15~25℃,滴加4ml 6n盐酸水溶液,反应完全后静置分层,有机相用饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤,有机相干燥,浓缩,得到1.1g固体状的化合物7-ii-1,收率为78%。
[0056]
第二步:将化合物7-ii-1(0.7g,1.4mmol)溶于乙醇(10ml)中,室温下搅拌加入氢氧化钠水溶液(0.17g,4.2mmol),tlc监测反应,反应完毕后减压浓缩,加入2n盐酸水溶液调节ph至1~2,用乙酸乙酯(50ml
×
2)萃取,合并有机层,无水硫酸钠干燥,浓缩,得到0.46g类白色固体状的化合物7-ii,收率为70%。
[0057]
第三步:将化合物7-ii(0.4g,0.9mmol)溶于二氯甲烷(5ml),分别加入化合物iii(0.26g,0.9mmol)和dmap(4-二甲氨基吡啶,0.12g,1mmol)并降温至0℃,加入dcc(n,n'-二环己基碳酰亚胺,0.2g,1mmol),缓慢升至室温,继续反应2小时,tlc监测反应,反应完成后,加入乙酸乙酯,过滤得滤液,滤液减压浓缩得油状物,经柱层析,得到0.3g固体状的化合物7,收率为44.0%,esi( )m/z=733.2[m h]
。
[0058]
实施例8
化合物8的结构式如式8所示,其为类白色固体,esi( )m/z=716.8[m h]
。
[0059]
《生物学评价试验》u87mg human glioblastoma(atcc)3h thymidine incorporation protocol生长培养基:含有earle salts的brl基本必需培养基(500ml) 5ml brl men 非必要氨基酸(10mm) 5ml brl青霉素和链霉素(10000微克/ml,10000微克/ml) 5ml brl丙酮酸钠溶液(100mm) 5ml brl l-谷氨酸盐(200mm) 50ml胎牛血清试验步骤如下:1、细胞受胰蛋白酶作用,以200微升生长培养基的最终体积以104细胞/孔的浓度放入96孔平底板中,在37℃粘连24小时。
[0060]
2、用移液管仔细去除培养基以不扰乱细胞单层,在每个孔中加入200微升新鲜生长培养基,分别加入10微升化合物1~8,在37℃再培养48小时。
[0061]
3、在培养最后5小时,板用1μ ci 3
h-胸腺嘧啶脱氧核苷标定。加入10μl 化合物1~8中的11μ ci,板放回培养器中培养最后5小时。
[0062]
4、用移液管仔细去除放射性培养基以不扰乱细胞单层。在每个孔中加入50 μl brl 10x胰蛋白酶,随后在37℃下培养10分钟或直至单层由底孔脱落。使用skatron96孔收获器在玻璃纤维过滤垫上收获样品,在wallac betaplate计数器上计数。计算得到目标化合物的ic
50
值见表1:表1由表1可知,化合物1-8均对mtor有良好的抑制作用,其中,化合物1、6抑制活性最高。
[0063]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。