1.本发明属于医药技术领域,尤其涉及一种一氧化氮纳米前药及其制备方法与应用。
背景技术:
2.尽管癌症治疗取得了显著进展,但癌症仍是威胁人类健康的重大疾病之一。光动力疗法(pdt)是一种微创治疗方式,对肿瘤细胞具有选择性细胞毒性。pdt基于光活化光敏剂(pss),在特定激光照射下将能量传递给组织氧,产生细胞毒性活性氧(ros),从而杀死肿瘤细胞而不损害周围的健康组织。
3.然而,光敏剂光照后产生的ros浓度显着影响pdt的疗效,由于许多因素会极大地影响ros浓度,光动力治疗临床应用受到很大限制。首要问题是癌细胞内谷胱甘肽(gsh)水平高,几乎是正常细胞的四倍。gsh是一种细胞内抗氧化剂,可有效清除产生的ros,从而降低pdt功效。gsh水平的下调可增强ros的产生,是提高pdt治疗效率的一种有前景的策略。其次,由于血管系统异常和肿瘤增殖活性的增加导致肿瘤缺氧微环境。氧气对于pdt的应用至关重要,缺氧直接削弱了pdt的抗肿瘤效率。此外,pdt过程中会消耗大量o2,加重肿瘤缺氧。
4.能同步消耗gsh和缓解肿瘤缺氧的策略是增强pdt的新兴策略。但只有少数研究人员致力于开发新型多功能分子一氧化氮(no)系统实现将gsh消耗和缺氧缓解相结合。研究表明,no显着抑制呼吸并影响各种生理和病理过程,包括癌症代谢等,作为心血管系统中的血管扩张剂,no通过松弛平滑肌细胞(smcs)缓解肿瘤缺氧和增加血管灌注。no还通过影响其在细胞中的代谢催化加速gsh/gssg氧化还原反应。
技术实现要素:
5.本发明的目的在于克服上述现有技术的不足之处而提供一种一氧化氮纳米前药及其制备方法与应用。
6.为实现上述目的,本发明采取的技术方案为:一氧化氮纳米前药,所述一氧化氮纳米前药为脱镁叶绿酸盐a、n-乙酰基-3-亚硝基硫醇-d-缬氨酸和聚乙二醇2000-羧基共修饰的pamam聚合物。
7.本发明的技术方案提供的一种一氧化氮纳米前药可以通过增强渗透性和保留(epr)在肿瘤中积累,当处于肿瘤中高谷胱甘肽(gsh)环境时,no纳米前药被激活释放一氧化氮,同时消耗gsh,从而舒张血管,减少肿瘤缺氧;由于gsh消耗和氧气供应增加,用660nm激光照射会触发光敏剂脱镁叶绿酸盐a(pa)产生更多的细胞毒性活性氧(ros),从而增强光动力疗法(pdt);本发明提供的一氧化氮纳米前药可解决肿瘤微环境引起的限制pdt的两个因素,因此可应用于临床肿瘤治疗上。
8.作为本发明所述一氧化氮纳米前药的优选实施方式,所述pamam为第四代pamam,其氨基个数为64个,毒性相对五代较低,而活性基团数又优于三代,便于修饰其他药物材料。
9.另外,本发明还提供了所述一氧化氮纳米前药的制备方法,包括以下步骤:
10.(1)pamam-pa的合成:脱镁叶绿酸盐a与pamam上的游离氨基发生酰胺化反应形成pamam-pa;
11.(2)n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷的合成:n-乙酰基-d-青霉胺与乙酸酐和吡啶发生反应形成硫内酯,得n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷;
12.(3)pamam-pa/sh的合成:n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷开环并与pamam-pa上的游离氨基反应生成pamam-pa/sh;
13.(4)pamam-pa/sno的合成:pamam-pa/sh的巯基上的氢被亚硝基取代形成pamam-pa/sno;
14.(5)一氧化氮纳米前药的合成:pamam-pa/sno上游离的氨基与聚乙二醇2000-羧基发生酰胺化反应后纯化得一氧化氮纳米前药。
15.作为本发明所述制备方法的优选实施方式,所述步骤(1)中,发生酰胺化反应形成pamam-pa包括以下步骤:将脱镁叶绿酸盐a、n-羟基琥珀酰亚胺和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺加入到二甲基亚砜中,并于室温下搅拌2-4h,接着加入pamam在室温下继续搅拌反应18-24h,反应结束后,透析、冷冻干燥,得pamam-pa;
16.优选地,发生酰胺化反应形成pamam-pa包括以下步骤:将脱镁叶绿酸盐a、n-羟基琥珀酰亚胺和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺加入到二甲基亚砜中,并于室温下搅拌2h,接着加入pamam在室温下继续搅拌反应24h,反应结束后,透析、冷冻干燥,得pamam-pa;
17.优选地,所述脱镁叶绿酸盐a、n-羟基琥珀酰亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺和pamam的质量比为脱镁叶绿酸盐a:n-羟基琥珀酰亚胺:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺:pamam=1:(1-2):(1-2):(0.05-0.1);
18.优选地,所述脱镁叶绿酸盐a、n-羟基琥珀酰亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺和pamam的质量比为脱镁叶绿酸盐a:n-羟基琥珀酰亚胺:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺:pamam=1:1.5:1.5:0.067;
19.优选地,所述透析使用的透析袋为mwco 3500,透析采用的溶剂为纯水,透析的时间为24h。
20.作为本发明所述制备方法的优选实施方式,所述步骤(2)中,发生反应形成硫内酯得n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷包括以下步骤:将n-乙酰基-d-青霉胺溶解于吡啶中并维持在0℃,接着将温度为0℃的乙酸酐/吡啶的混合溶液滴加进n-乙酰基-d-青霉胺的吡啶溶液中,冰浴反应1h后转移至室温下搅拌反应过夜,反应结束后,除去吡啶,得不溶物,将不溶物溶解于氯仿中,并用1m盐酸洗涤3次,收集有机层并用硫酸镁干燥,然后过滤得滤液,浓缩滤液得固体,将所得固体溶解于正己烷中并结晶,所得晶体为n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷;
21.优选地,所述n-乙酰基-d-青霉胺、吡啶和乙酸酐的质量体积比为n-乙酰基-d-青霉胺:吡啶:乙酸酐=(0.1-1)g:1g:1ml;
22.优选地,所述n-乙酰基-d-青霉胺、吡啶和乙酸酐的质量体积比为n-乙酰基-d-青霉胺:吡啶:乙酸酐=0.3g:1g:1ml。
23.作为本发明所述制备方法的优选实施方式,所述步骤(3)中,反应生成pamam-pa/
sh包括以下步骤:将pamam-pa和n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷溶解于二甲基亚砜中,室温搅拌20-24h,接着透析、冷冻干燥,得pamam-pa/sh;
24.优选地,反应生成pamam-pa/sh包括以下步骤:将pamam-pa和n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷溶解于二甲基亚砜中,室温搅拌24h,接着透析、冷冻干燥,得pamam-pa/sh;
25.优选地,pamam-pa和n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷的摩尔质量比为(4-5):1,pamam-pa与二甲基亚砜的质量体积比为(6-6.5)mg:1ml;
26.优选地,pamam-pa和n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷的摩尔质量比为4.5:1,pamam-pa与二甲基亚砜的质量体积比为6mg:1ml;
27.优选地,所述透析使用的透析袋为mwco 3500,透析采用的溶剂为纯水,透析的时间为24h。
28.作为本发明所述制备方法的优选实施方式,所述步骤(4)中,形成pamam-pa/sno包括以下步骤:将亚硝酸叔丁酯与pamam-pa/sh混合,于0℃下避光搅拌过夜后,超滤、冷冻干燥,得pamam-pa/sno;其中,pamam-pa/sh与亚硝酸叔丁酯的摩尔质量比为(3.5-4):1;
29.优选地,形成pamam-pa/sno包括以下步骤:将亚硝酸叔丁酯与pamam-pa/sh混合,于0℃下避光搅拌过夜后,超滤、冷冻干燥,得pamam-pa/sno;其中,pamam-pa/sh与亚硝酸叔丁酯的摩尔质量比为3.75:1;
30.优选地,所述超滤采用的溶剂为超纯水,超滤采用的超滤膜为mwco 10000da。
31.作为本发明所述制备方法的优选实施方式,所述步骤(5)中,发生酰胺化反应得一氧化氮纳米前药包括以下步骤:将聚乙二醇2000-羧基溶于二甲基亚砜,接着加入n-羟基琥珀酰亚胺和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺,室温搅拌2h,接着加入pamam-pa/sno室温下搅拌24h,反应结束后,将反应混合物加入到超纯水中后超滤、冷冻干燥,得一氧化氮纳米前药;
32.优选地,聚乙二醇2000-羧基、n-羟基琥珀酰亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺和pamam-pa/sno的摩尔质量比为聚乙二醇2000-羧基:n-羟基琥珀酰亚胺:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺:pamam-pa/sno=1:(0.0286-0.0333):(0.0500-0.0667):(1-10);
33.优选地,聚乙二醇2000-羧基、n-羟基琥珀酰亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺和pamam-pa/sno的摩尔质量比为聚乙二醇2000-羧基:n-羟基琥珀酰亚胺:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺:pamam-pa/sno=1:0.0303:0.526:5;
34.优选地,所述聚乙二醇2000-羧基与二甲基亚砜的质量体积比为(2.5-3)mg:1ml;
35.优选地,所述聚乙二醇2000-羧基与二甲基亚砜的质量体积比为2.77mg:1ml;
36.优选地,所述超滤采用的溶剂为超纯水,超滤采用的超滤膜为mwco 50000da;
37.优选地,所述冷冻干燥后进一步通过纳米沉淀法处理,得一氧化氮纳米前药。
38.另外,本发明还提供了所述一氧化氮纳米前药在制备抗肿瘤药物上的应用。
39.与现有技术相比,本发明的有益效果为:
40.(1)本发明提供的一氧化氮纳米前药能通过消耗肿瘤区域谷胱甘肽,降低谷胱甘肽对活性氧产生的抑制作用,从而增强随后的光动力治疗的疗效;
41.(2)本发明提供的一氧化氮纳米前药具有肿瘤细胞的选择性,降低对正常细胞的
损伤,还可以通过自身荧光来检测其在肿瘤组织和细胞摄取情况;
42.(3)本发明提供的一氧化氮纳米前药可以通过释放一氧化氮,舒张肿瘤区域血管,缓解肿瘤缺氧情况,从而缓解肿瘤缺氧对后续光动力治疗效果的抑制作用;
43.(4)本发明提供的一氧化氮纳米前药,可以通过自身荧光检测药物在肿瘤组织或细胞富集与摄取情况;可以协同缺氧缓解及谷胱甘肽下降,显著提升光动力治疗效果,极大提升诱导肿瘤细胞凋亡能力;
44.(5)本发明提供的一氧化氮纳米前药的制备方法简单易操作,有利于大量合成。
附图说明
45.图1为本发明实施例1中一氧化氮纳米前药np
pa/no
的合成路线图;
46.图2为实施例1制备得到的np
pa/no
的核磁共振氢谱图;
47.图3为效果例1中不同浓度np
pa/no
处理后的一氧化氮释放曲线图;
48.图4为效果例1中不同浓度np
pa/no
处理后的谷胱甘肽响应紫外波谱变化曲线图;
49.图5为效果例1中660nm激光照射后np
pa/no
产生活性氧的紫外波谱变化曲线图;
50.图6为效果例2中激光共聚焦观察细胞对np
pa/no
的摄取的荧光定量图;
51.图7为效果例2中np
pa/no
在660nm光照后细胞内活性氧产生示意图;
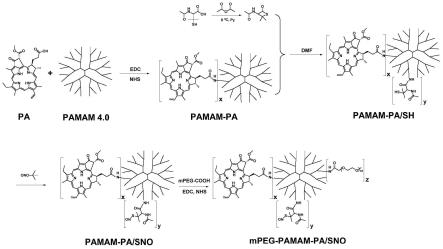
52.图8为效果例2中np
pa/no
处理后谷胱甘肽水平变化图;
53.图9为效果例2中np
pa/no
处理后一氧化氮水平变化图;
54.图10为效果例2中np
pa/no
与不同对照组处理后细胞流式凋亡图;
55.图11为效果例2中np
pa/no
光照处理前后的活死细胞染色图;
56.图12为效果例3中np
pa/no
体内生物分布图;
57.图13为效果例3中np
pa/no
缓解乏氧的荧光定量图;
58.图14为效果例3中np
pa/no
治疗后肿瘤生长抑制曲线图;
59.图15为效果例3中np
pa/no
治疗后小鼠体重变化曲线图;
60.图16为效果例3中np
pa/no
治疗后小鼠肿瘤解剖图。
具体实施方式
61.为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。
62.实施例1
63.1、本发明实施例的一氧化氮纳米前药np
pa/no
的制备,制备路线图如图1所示,具体包括以下步骤:
64.(1)pamam-pa的合成:将脱镁叶绿酸盐a(pa,62.55mg,105.53μmol)、n-羟基琥珀酰亚胺(nhs)(18.20mg,158.30μmol)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(edc)(30.39mg,158.30μmol)混合物在dmso(5ml)中室温搅拌2小时;然后加入pamam 4.0(100mg,7.04μmol)并在室温下继续反应24小时;反应结束后,用透析袋(mwco 3500)将溶液在纯水中透析24小时。冷冻干燥后得到产品pamam-pa;
65.(2)n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷(snap)的合成:将n-乙酰基-d-青霉胺(1.5g,7.84mmol)溶解在吡啶(5ml)中冰浴反应30分钟;然后,将与5ml乙酸酐混合的
5ml吡啶在冰上再冷却30分钟;之后,将混合溶液滴加到上述吡啶溶液中,冰浴反应1小时,室温搅拌过夜;然后通过旋转蒸发除去溶剂;将粗品溶解于氯仿中,用1m盐酸洗涤3次;收集有机层并用硫酸镁干燥。然后过滤固体,同时旋转蒸发所得溶液以除去溶剂;剩余固体在己烷中结晶并在真空过滤下干燥以获得n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷;
66.(3)pamam-pa/sh的合成:将pamam-pa(30mg,1.29μmol)和n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷(6.69mg,38.7μmol)溶解在5ml dmso中,并在室温下搅拌24小时;然后将混合物用透析袋(mwco 3500)在纯水中透析24小时并冷冻干燥以获得pamam-pa/sh;
67.(4)pamam-pa/sno的合成:往pamam-pa/sh中加入亚硝酸叔丁酯(8.01mg,77.4μmol),搅拌混合溶液,0℃避光过夜搅拌;反应后将溶液加入超纯水中并通过超滤(mwco 10000da)进一步纯化以去除杂质;接着真空冷冻干燥后得到pamam-pa/sno;
68.(5)一氧化氮纳米前药(mpeg-pamam-pa/sno)的合成:将peg2000-cooh(5.54mg,2.77μmol)溶于2ml dmso,然后加入edc(0.29mg,3.3μmol)和nhs(0.17mg,3.3μmol),室温活化2h,然后加入pamam-pa/sno(25.82mg,0.92μmol),混合溶液在室温下搅拌24h;之后将混合物加入超纯水中以获得纳米颗粒,并通过超滤(mwco 50000da)进一步纯化以去除杂质;真空冷冻干燥后得到mpeg-pamam-pa/sno;
69.(6)一氧化氮纳米前药(mpeg-pamam-pa/sno)的纯化:采用纳米沉淀法对mpeg-pamam-pa/sno进行进一步纯化,将mpeg-pamam-pa/sno溶解在1.0ml的dmso溶液中,然后在搅拌下逐渐加入10ml超纯水;在搅拌2小时后,将溶液转移到透析袋(mwco 3500)中,用超纯水去除dmso得纯化后的一氧化氮纳米前药,标记为np
pa/no
;
70.图1展示了本发明实施例1的制备过程,其中图1中的x、y、z分别表示脱镁叶绿酸盐a、n-乙酰基-2,2-二甲基-4-氧代-3-硫杂环丁烷和聚乙醇2000-羧基对pamam 4.0的修饰量,pamam 4.0采用简易的画法表示其树枝状的结构,树枝的端部表示具有反应活性的游离氨基,x、y、z即分别表示上述三者的羧基与pamam 4.0上的游离氨基的反应量,其中,x y z≤64。
71.2、核磁表征:
72.对制备得到的一氧化氮纳米前药np
pa/no
进行核磁表征,表征如图2所示,从图2可以看出,本发明实施例1成功的制备得到了一氧化氮纳米前药np
pa/no
。
73.实施例2
74.本实施例为np
pa
的合成:所述np
pa
为脱镁叶绿酸盐a和聚乙二醇2000-羧基共修饰的pamam聚合物;可用于后续np
pa/no
效果验证时的对照物;
75.(1)pamam-pa的合成:将脱镁叶绿酸盐a(pa,62.55mg,105.53μmol)、n-羟基琥珀酰亚胺(nhs)(18.20mg,158.30μmol)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(edc)(30.39mg,158.30μmol)混合物在dmso(5ml)中室温搅拌2小时;然后加入pamam 5.0(100mg,7.04μmol)并在室温下继续反应24小时;反应结束后,用透析袋(mwco 3500)将溶液在纯水中透析24小时。冷冻干燥后得到产品pamam-pa;
76.(2)一氧化氮纳米前药(mpeg-pamam-pa/sno)的合成:将peg2000-cooh(5.54mg,2.77μmol)溶于2ml dmso,然后加入edc(0.29mg,3.3μmol)和nhs(0.17mg,3.3μmol),室温活化2h,然后加入pamam-pa(25.82mg,0.92μmol),混合溶液在室温下搅拌24h;之后将混合物加入超纯水中以获得纳米颗粒,并通过超滤(mwco 50000da)进一步纯化以去除杂质;真空
冷冻干燥后得到mpeg-pamam-pa;
77.(3)mpeg-pamam-pa的纯化:采用纳米沉淀法对mpeg-pamam-pa进行纯化,将mpeg-pamam-pa溶解在1.0ml的dmso溶液中,然后在搅拌下逐渐加入10ml超纯水;在搅拌2小时后,将溶液转移到透析袋(mwco 3500)中,用超纯水去除dmso得纯化后的mpeg-pamam-pa,标记为np
pa
。
78.效果例1
79.对实施例1制备得到的一氧化氮纳米前药np
pa/no
进行体外性能的测试,具体测试项目及结果分别记录如下。
80.1、一氧化氮纳米前药np
pa/no
在不同的gsh浓度下一氧化氮的释放
81.设置三个不同梯度浓度的gsh,分别为0μm、5μm、10mm,然后加入相同质量的一氧化氮纳米前药np
pa/no
,在相同的条件下测试不同gsh浓度的体系下no释放量随时间的变化量;从图3中可以看出,np
pa/no
在体外具有良好的gsh响应并释放no的能力。
82.2、不同浓度的一氧化氮纳米前药np
pa/no
与gsh反应后gsh紫外吸收的变化
83.设置三个不同梯度浓度的np
pa/no
,分别为5μg/ml、10μg/ml、20μg/ml,同时设置一个空白对照以及只添加gsh的对照,从图4中可以看出,np
pa/no
在体外具有良好的gsh消耗能力。
84.3、一氧化氮纳米前药np
pa/no
体外活性氧生成能力测试
85.为了研究np
pa/no
的ros生成能力,将9,10-蒽二基-双(亚甲基)二丙二酸(abda)加入到np
pa/no
溶液中,abda与单线态氧反应吸光度会降低;其中,使用激光的波长为660nm,功率为0.30w*cm-2
,照射不同时间后通过uv-vis进行测量;从测量结果图5中可以看出,np
pa/no
在体外具有良好的活性氧生成能力。
86.效果例2
87.对实施例1制备得到的一氧化氮纳米前药np
pa/no
进行体外细胞实验,具体测试项目及结果如下:
88.1、肿瘤细胞对np
pa/no
的摄取
89.选取小鼠乳腺癌4t1细胞系测试细胞对np
pa/no
的摄取;使用clsm观察4t1细胞对np
pa/no
的摄取,将4t1细胞以5
×
104个细胞的密度接种于35mm玻璃底培养皿中并孵育过夜;然后弃去培养基,将细胞与含有np
pa/no
(pa浓度10μg*ml-1
)的新鲜培养基一起孵育2小时或4小时;然后用pbs洗涤细胞并用hoechst 33342标记10分钟,最后用pbs洗涤细胞2次,clsm观察pa和hoechst 33342的荧光;测试结果如图6所示,从图6中可以看出,随着孵育时间的延长,细胞内np
pa/no
的荧光信号逐渐增强,表明np
pa/no
可以被细胞摄取。
90.2、np
pa/no
光照后在肿瘤细胞内活性氧产生
91.以dcfh-da为指示剂检测细胞内ros的产生,在ros存在下,dcfh-da可被氧化产生荧光化合物dcf;将4t1细胞以5
×
104个细胞的密度接种到35mm玻璃底培养皿中并孵育过夜;之后,弃去培养基,将细胞与含有np
pa/no
(pa浓度10μg*ml-1
)的新鲜培养基孵育4小时;随后,将细胞用pbs洗涤3次并用dcfh-da染色;孵育20分钟后,用pbs洗涤细胞并用功率0.30w*cm-2
的660nm激光照射5分钟,然后用pbs洗涤细胞并用hoechst 33342标记10分钟,用clsm观察dcf的荧光强度;测试结果如图7所示,从图7中可以看出,np
pa/no
在660nm激光照射后具有良好的细胞内活性氧产生效果。
92.3、np
pa/no
消耗肿瘤细胞内谷胱甘肽能力
93.使用gsh探针(thioltracker
tm
violet)研究纳米颗粒降低细胞内gsh的效果;将4t1乳腺癌细胞接种到96孔荧光板中并孵育过夜,然后弃去培养基,用np
pa/no
(pa浓度10μg*ml-1
)处理细胞,4小时后,用pbs洗涤细胞,并根据gsh探针说明书检测流程,使用biotek cytation 5cell imaging multi-mode reader分析细胞内gsh水平,测试结果如图8所示,从图8中可以看出,相对pbs组,加入np
pa
组gsh水平几乎不变,而加入np
pa/no
组具有良好的细胞内谷胱甘肽消耗能力,其消耗gsh效果与gsh消耗剂bso类似。
94.4、np
pa/no
在肿瘤细胞内产生一氧化氮能力
95.3-氨基,4-氨基甲基-2',7'-双荧光素二乙酸酯(daf-fm da)作为no荧光指示剂以检测4t1细胞中的no;将4t1乳腺癌细胞接种到96孔板中并培养24小,然后弃去培养基,使用daf-fm da孵育20min,然后用pbs洗涤3次,再用np
pa/no
(pa浓度10μg*ml-1
)孵育细胞,4小时后,用pbs洗涤细胞并根据no探针说明书流程,使用biotek cytation 5cell imaging multi-mode reader分析细胞内no水平,测试结果如图9所示;从图9中可以看出,加入np
pa
组与pbs组荧光强度类似,而np
pa/no
组no荧光明显增高,而使用gsh消耗剂bso之后no荧光有所下降,说明是由于gsh引起的no产生,并且np
pa/no
具有良好的细胞内一氧化氮产生能力。
96.5、np
pa/no
在肿瘤细胞内诱导细胞凋亡效果
97.采用annexin v-fitc/pi细胞凋亡检测试剂盒进一步研究pdt诱导的细胞凋亡效应;将4t1细胞以3
×
104/孔的密度接种于24孔板中孵育过夜,加入含有不同浓度np
pa/no
的培养基培养4小时,之后,弃去培养基,将细胞与新鲜培养基一起孵育,然后在功率0.30w*cm-2
的波长660nm激光照射5分钟,再孵育18小时后,收集细胞并用pbs洗涤两次,然后用annexin v和pi处理,最后,通过流式细胞术分析(bd facscelesta,san jose,ca)测量细胞,检测结果如图10所示;从图10中可以看出,使用膜联蛋白v fitc/pi细胞凋亡分析对pdt有效性进行分析表明,辐照np
pa/no
处理的细胞导致45.7%的细胞凋亡率,而对照组np
pa
处理的细胞为22.4%,未辐照的细胞凋亡率为10%。
98.6、np
pa/no
光照后杀伤肿瘤细胞效果
99.将4t1细胞以1
×
105/孔的密度接种到35mm玻璃底培养皿中并孵育过夜,加入含有不同浓度np
pa/no
的培养基并孵育4小时,之后,弃去培养基,将细胞与新鲜培养基一起孵育,然后在功率0.30w*cm-2
的波长660nm激光照射5分钟,再孵育18小时后,使用活死染色试剂盒(bestbio science,china)研究细胞活力,染料钙黄绿素am将活细胞染成绿色,碘化丙啶将死细胞染成红色,使用clsm观察活细胞或死细胞的荧光,测试结果如图11所示;从图11中可以看出,在没有照射的情况下显示出最小的细胞毒性,而在660nm激光照射(5分钟,0.30w*cm-2
)下,np
pa/no
相对于np
pa
引发了显着更高的细胞毒性。
100.效果例3
101.对实施例1制备得到的一氧化氮纳米前药np
pa/no
进行动物水平实验,具体测试项目及结果如下:
102.1、活体肿瘤荧光成像实验
103.使用in-vivo xtreme(bruker,德国)对4t1荷瘤babl/c裸鼠进行np
pa/no
的实时成像;裸鼠右侧乳腺注射5
×
10
6 4t1细胞,肿瘤体积达到150mm3后,将荷瘤小鼠随机分为3组,分别静脉注射100μl pbs、np
pa
和np
pa/no
,pa剂量为3mg*kg-1
,在注射后4、8、24或48小时收集小鼠的荧光图像,在注射后48小时处死小鼠,从小鼠身上采集肿瘤和主要器官并进行离体
成像;从图12中可以看出,np
pa/no
在肿瘤部位的积累在长达48小时内相对稳定,并且肿瘤部位的荧光强度以时间依赖性方式变化,在注射后8小时左右达到最大强度,np
pa/no
治疗的小鼠在肿瘤部位表现出更强的荧光强度,归因于纳米平台的epr效应。
104.2、缺氧染色的肿瘤切片的免疫荧光图像
105.用低氧探针(盐酸哌莫硝唑)进一步研究了np
pa/no
的缺氧缓解效果,荷瘤小鼠以pa浓度5mg*kg-1
的剂量注射pbs、np
pa
和np
pa/no
,然后将探针以60mg*kg-1
的剂量静脉注射到携带4t1的小鼠中,90分钟后处死,然后在冷冻条件下切除肿瘤切片,然后按照hp6-hypoxyprobetm green kit(hypoxyprobe,inc.,usa)的说明,将冷冻的肿瘤切片用fitc标记的hypoxyprobe抗体染色以及4',6-二脒基-2'-苯基吲哚(dapi)染色,然后通过clsm观察,测试结果如图13所示;从图13中可以看出,蓝色荧光代表细胞核,绿色荧光代表肿瘤缺氧区域,该分析显示,np
pa/no
治疗的肿瘤中绿色荧光显着降低,表明与pbs治疗的肿瘤相比,缺氧阳性区域显着减少。
106.3、体内治疗实验
107.当肿瘤体积达到约150mm3时,将4t1荷瘤小鼠随机分为5组,分别给小鼠静脉注射pbs、np
pa
和np
pa/no
,pa剂量为5mg*kg-1
,用660nm波长激光以0.30w*cm-2
功率局部照射20min,8h后,没有激光的小鼠作为对照,隔日测量肿瘤体积和体重,计算肿瘤体积的公式为:v=l
×w×
w/2(l,最长尺寸;w,最短尺寸),在实验结束14天时将小鼠处死,收集肿瘤拍照分析,实验结果如图14-16所示;从图14-图16中可以看出,在没有照射的情况下,用pbs和np
pa
处理的组中肿瘤生长最快,在没有激光照射的np
pa/no
治疗组和np
pa
加照射治疗组中,肿瘤生长受到轻度抑制,表明治疗效果不明显,然而,在用np
pa/no
加照射治疗的组中,肿瘤生长受到强烈抑制,在不同组的小鼠中没有观察到显着的体重差异,表明np
pa/no
具有良好的生物相容性和最小的毒性,在第14天收集肿瘤和其他器官并称重肿瘤,解剖肿瘤的照片表明np
pa/no
与激光的pdt效果很好,这与上述结果相似。
108.最后应当说明的是,以上实施例以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。