毛萼内酯素a和毛萼内酯素b及其制备方法
技术领域
1.本发明属于天然产物有机合成技术领域和药物技术领域,具体涉及一种新型对映-贝壳杉烷二萜类化合物毛萼内酯素a和毛萼内酯素b、药物组合物及其制备方法。
背景技术:
2.天然产物骨架为活性分子的药物化学研究提供了可借鉴的模板。例如天然螺环内酯型以及延命素型对映-贝壳杉烷型二萜为冬凌草甲素(oridonin)的结构修饰提供了借鉴。毛萼内酯素a是从毛萼香茶菜(isodon eriocalyx)中发现的具有新颖的1,10裂环-2,20-环化-1,20内酯化-对映-贝壳杉烷型骨架。由于毛萼内酯素a的样品量较少,无法确定其绝对构型和开展生物活性研究。本发明希望能够通过半合成手段得到毛萼内酯素a,在解决绝对构型及样品量问题的同时,能够给oridonin提供新的结构修饰方法,基于oridonin得到结构修饰前体分子毛萼内酯素b。
3.目前,现有技术中未见有毛萼内酯素a与毛萼内酯素b制备方法的报道。
技术实现要素:
4.本发明的目的是提供一种新型对映-贝壳杉烷二萜类化合物毛萼内酯素a和毛萼内酯素b,以其为活性成分的药物组合物和它们的制备方法。本发明将基于分子内沃夫重排串联内酯化反应构建关键的新型骨架,完成毛萼内酯素a与毛萼内酯素b的制备,具有非常重要的方法学意义、研究价值和应用前景。
5.为实现本发明的上述目的,本发明提供了如下的具体方案:
6.如下结构式所示的一种新型对映-贝壳杉烷型二萜化合物毛萼内酯素a,以及新型结构修饰前体化合物毛萼内酯素b,
[0007][0008][0009]
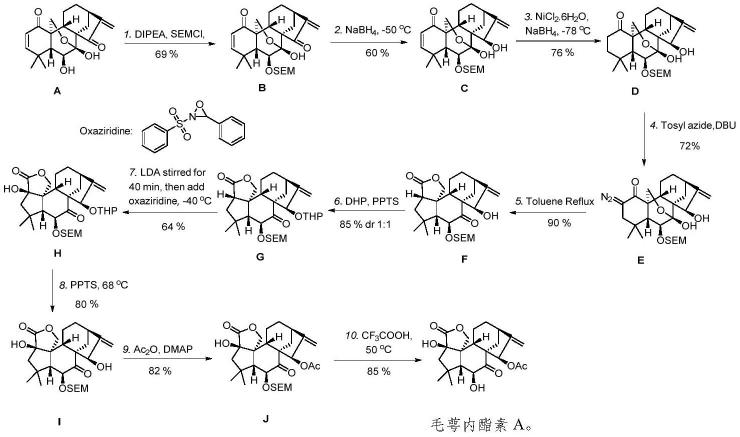
本发明同时提供了所述的新型对映-贝壳杉烷二萜类化合物毛萼内酯素a的制备方法,该方法以毛萼乙素(a)为起始原料先通过四步反应得到中间体e,中间体e通过关键的分子内沃夫重排串联内酯化反应(5)构建关键中间体,再经过五步反应得到毛萼内酯素a,其简化流程如下:
[0010][0011]
所述的毛萼内酯素a的制备方法,该方法流程如下:
[0012][0013]
如所述的毛萼内酯素a的制备方法,该方法包括如下步骤:
[0014]
(1)制备中间体b:将化合物a(0.01~100g)溶解在(1~100ml)的二氯甲烷中,加入二异丙基乙胺(1~100ml)。在0℃冰浴下缓慢滴加sem-cl(1~100ml)。然后将反应置于室温中反应4个小时,tlc检测反应完全后,饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤、无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体b。
[0015][0016]
(2)制备中间体c:将中间体b溶解在(1~100ml)的四氢呋喃中,置于
–
50℃的低温
反应器中充分冷却15分钟。后缓慢加入nabh4(1~15g),充分反应30分钟。tlc检测反应完全后,使用丙酮淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体b。
[0017][0018]
(3)制备中间体d:将上述所制备c溶解在(1~100ml)的甲醇中,先加入六水合二氯化镍充分搅拌半小时;后置于
–
78℃的低温反应器中充分冷却半个小时。分三次缓慢加入nabh4(1~15g),加入后反应液立刻变黑,充分反应1小时后,将反应移至室温继续反应1个小时。tlc检测反应完全后,饱和nahco3淬灭,淬灭后加入(1~100g)硅藻土,搅拌均匀后分别使用etoac抽滤,抽滤后加水萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体d。
[0019][0020]
(4)制备中间体e:将中间体d溶解在(1~100ml)的乙腈中,依次加入dbu(1~20ml)和对甲基苯磺酰氯叠氮(1~20ml)。该反应置于室温中充分反应2小时。tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体e。
[0021][0022]
(5)制备中间体f:将化合物e溶解于(1~10ml)甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,得到无定型白色固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体f。
[0023][0024]
(6)制备中间体g:将化合物f溶解在(1~10ml)的二氯甲烷中,反应置于室温中依次添加dhp(1~10ml)与对甲苯磺酸吡啶盐(0.5~5g)。室温充分反应15分钟,使用tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体g。
[0025][0026]
(7)制备中间体h:将化合物g溶解在(1~10ml)的四氢呋喃中,反应置于
–
40℃的低温反应器中充分冷却10分钟后,先添加lda(1~10ml)与底物反应40分钟。后加入oxaziridine(1~10g),
–
40℃继续充分反应2个小时,使用tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体h。
[0027][0028]
(8)制备中间体i:将化合物h溶解在(1~10ml)的乙醇中,添加对甲苯磺酸吡啶盐(0.1~5g)在68℃中反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体i。
[0029][0030]
(9)制备中间体j:将化合物i溶解在(1~10ml)的二氯甲烷中,在冰浴条件下依次加入乙酸酐(0.1~1ml),dmap(0.1~1g)中反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体j。
[0031][0032]
(10)制备毛萼内酯素a:将化合物j溶解在1ml的四氢呋喃中,在50℃加热条件下添加三氟乙酸(0.1~1ml)反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为目标化合物毛萼内酯素a。
[0033][0034]
其中流程中所述的分子内沃夫重排串联内酯化反应(5)步骤包括以下条件:
[0035][0036]
note:alsolated yield after flash chromatography;babbreviations:blue led,blue light emitting diode;hpsl,high pressure sodium lamp;hpml,high pressure mercury lamp.
[0037]
所述的分子内沃夫重排串联内酯化反应(5)用于新型冬凌草甲素衍生物的制备。该方法流程如下:
[0038][0039]
毛萼内酯素b的制备方法,该方法包括如下步骤:
[0040]
(1)制备中间体l:将化合物k溶解在(1~100ml)的丙酮中,在0℃冰浴条件下添加琼斯试剂(10ml)反应15分钟,使用tlc检测反应完全后,使用异丙醇淬灭,加水稀释后etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物l,经质谱、一维和二维核磁共振鉴定化合物为中间体l。
[0041][0042]
(2)制备中间体m:将化合物l溶解在(1~100ml)的1,2-二氯乙烷中,在0℃冰浴条件下加入nabh(oac)3(1~50g)后随后缓慢滴加乙酸(0.1~1ml)反应10分钟,使用tlc检测反应完全后,使用丙酮淬灭,加水稀释后etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体m。
[0043][0044]
(3)制备中间体n:将化合物m溶解在(1~100ml)的二氯甲烷中,在0℃冰浴条件下依次添加dipea(1~50ml)以及tmscl(1~50ml),在冰浴条件下反应30分钟后减压浓缩。浓缩后产品溶解于乙腈,后依次添加dbu(1~50ml)和对甲苯磺酰叠氮(1~50ml)反应2小时后滴加tbaf(1~50g)反应30分钟,使用tlc检测反应完全后,加水稀释加etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体n。
[0045][0046]
(4)制备中间体o:将化合物n溶解在(1~100ml)的二氯甲烷中,在0℃冰浴条件下依次添加nahco3(1~10g)以及戴斯-马丁试剂(1~10g),在冰浴条件下反应10分钟,使用tlc检测反应完全后,使用na2o3s2淬灭,加水稀释加etoac萃取,饱和食盐水、洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体o。
[0047][0048]
(5)制备毛萼内酯素b:将化合物o溶解于(1~50ml)甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,得到无定型白色固体化合物,经质谱、一维和二维核磁共振鉴定化合物为毛萼内酯素b。
[0049][0050]
毛萼内酯素a和/或毛萼内酯素b在制备抗肿瘤的药物中的应用,具体地,在制备抗宫颈癌、乳腺癌、结肠癌的药物中的应用。
[0051]
药物组合物,其包含所述的新型骨架类型化合物毛萼内酯素a和毛萼内酯素b,和至少一种药学上可接受的载体。
[0052]
药物组合物在制备抗宫颈癌、乳腺癌、结肠癌的药物中的应用。
[0053]
本发明新型对映-贝壳杉烷二萜类化合物毛萼内酯素a和毛萼内酯素b,用作药物时,可以直接使用,或者可以药物组合的形式使用。该药物组合物含有0.1~99%,优选为0.5~90%的本发明化合物,其余为药物学上可以接受的,对人和动物无毒和惰性的可药用载体或赋形剂。
[0054]
所述的可药用载体是一种或多种选自固体。半固体和液体稀释剂、超填料以及药物制品辅剂。将所述的有效提取物或有效部位以单位体重服用量的形式使用。本发明的药物可经口服和口腔喷雾两种形式给药。
[0055]
口服可用其固体或液体制剂,如粉剂、片剂、糖衣片剂、胶囊、酊剂、糖浆、滴丸剂等。
[0056]
口腔喷雾可用其固体或液体制剂。
[0057]
与现有技术相比,本发明具备如下的优异性:
[0058]
本发明新型对映-贝壳杉烷二萜类化合物毛萼内酯素a,是首次合成制备出来的,其绝对和相对构型通过核磁共振、质谱、圆二色谱(cd)、x单晶衍射等测定方法得以确定,命名为毛萼内酯素a,英文名为maoelactone a。它能通过本发明方法制备出来。以oridonin为原料,经沃夫重排串联内酯化反应构建结构修饰前体毛萼内酯素b。本发明的新型对映-贝壳杉烷二萜类化合物及合成中间体为首次合成,合成路线新颖,具备应用前景。
附图说明
[0059]
图1为新型对映-贝壳杉烷二萜类化合物毛萼内酯素a的合成路线流程图;
[0060]
图2为毛萼内酯素b的合成路线流程图;
[0061]
图3为化合物毛萼内酯素a和毛萼内酯素b的结构示意图。
具体实施方式:
[0062]
下面结合附图对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或改进,均落入本发明的保护范围
[0063]
实施例1
[0064]
本发明所制备的新型对映-贝壳杉烷二萜类化合物为从药用植物中分离得到的为毛萼内酯素a,英文名为maoelactone a。以毛萼乙素a为起始原料,通过化学反应实现对毛萼内酯素a的化学合成。又以oridonin为原料,经沃夫重排串联内酯化反应构建结构修饰前
体毛萼内酯素b。以上化合物结构式如下:
[0065][0066]
本发明的新型对映-贝壳杉烷二萜类化合物毛萼内酯素a及结构修饰前体毛萼内酯素b的制备方法为:
[0067]
(1)制备中间体b:将化合物a(0.01~100g)溶解在(1~100ml)的二氯甲烷中,加入二异丙基乙胺(1~100ml)。在0℃冰浴下缓慢滴加sem-cl(1~100ml)。然后将反应置于室温中反应4个小时,tlc检测反应完全后,饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤、无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体b。
[0068][0069]
(2)制备中间体c:将上诉所制备b溶解在(1~100ml)的四氢呋喃中,置于
–
50℃的低温反应器中充分冷却15分钟。后缓慢加入nabh4(1~15g),充分反应30分钟。tlc检测反应完全后,使用丙酮淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体b。
[0070][0071]
(3)制备中间体d:将上诉所制备c溶解在(1~100ml)的甲醇中,先加入六水合二氯化镍充分搅拌半小时;后置于
–
78℃的低温反应器中充分冷却半个小时。分三次缓慢加入nabh4(1~15g),加入后反应液立刻变黑,充分反应1小时后,将反应移至室温继续反应1个小时。tlc检测反应完全后,饱和nahco3淬灭,淬灭后加入(1~100g)硅藻土,搅拌均匀后分别使用etoac抽滤,抽滤后加水萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体d。
[0072][0073]
(4)制备中间体e:将化合物d溶解在(1~100ml)的乙腈中,依次加入dbu(1~20ml)和对甲基苯磺酰氯叠氮(1~20ml)。该反应置于室温中充分反应2小时。tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体e。
[0074][0075]
(5)制备中间体f:将化合物e溶解于(1~10ml)甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,得到无定型白色固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体f。
[0076][0077]
(6)制备中间体g:将化合物f溶解在(1~10ml)的二氯甲烷中,反应置于室温中依次添加dhp(1~10ml)与对甲苯磺酸吡啶盐(0.5~5g)。室温充分反应15分钟,使用tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体g。
[0078][0079]
(7)制备中间体h:将化合物g溶解在(1~10ml)的四氢呋喃中,反应置于
–
40℃的低温反应器中充分冷却10分钟后,先添加lda(1~10ml)与底物反应40分钟。后加入oxaziridine(1~10g),
–
40℃继续充分反应2个小时,使用tlc检测反应完全后,使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体h。
[0080][0081]
(8)制备中间体i:将化合物h溶解在(1~10ml)的乙醇中,添加对甲苯磺酸吡啶盐(0.1~5g)在68℃中反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到油状化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体i。
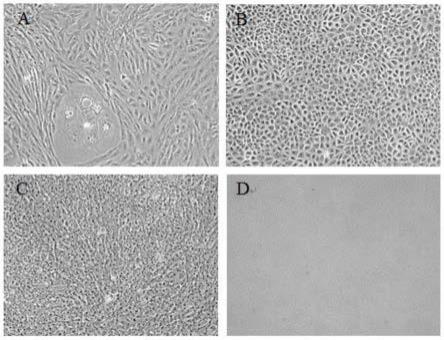
[0082][0083]
(9)制备中间体j:将化合物i溶解在(1~10ml)的二氯甲烷中,在冰浴条件下依次加入乙酸酐(0.1~1ml),dmap(0.1~1g)中反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体j。
[0084][0085]
(10)制备毛萼内酯素a:将化合物j溶解在1ml的四氢呋喃中,在50℃加热条件下添加三氟乙酸(0.1~1ml)反应1个小时。使用饱和nahco3淬灭,etoac萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为目标化合物毛萼内酯素a。
[0086][0087]
(11)制备中间体l:将化合物k溶解在(1~100ml)的丙酮中,在0℃冰浴条件下添加琼斯试剂(10ml)反应15分钟,使用tlc检测反应完全后,使用异丙醇淬灭,加水稀释后etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物l,经质谱、一维和二维核磁共振鉴定化合物为中间体l。
[0088][0089]
(12)制备中间体m:将化合物l溶解在(1~100ml)的1,2-二氯乙烷中,在0℃冰浴条件下加入nabh(oac)3(1~50g)后随后缓慢滴加乙酸(0.1~1ml)反应10分钟,使用tlc检测反应完全后,使用丙酮淬灭,加水稀释后etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体m。
[0090][0091]
(13)制备中间体n:将化合物m溶解在(1~100ml)的二氯甲烷中,在0℃冰浴条件下依次添加dipea(1~50ml)以及tmscl(1~50ml),在冰浴条件下反应30分钟后减压浓缩。浓缩后产品溶解于乙腈,后依次添加dbu(1~50ml)和对甲苯磺酰叠氮(1~50ml)反应2小时后滴加tbaf(1~50g)反应30分钟,使用tlc检测反应完全后,加水稀释加etoac萃取,饱和食盐水洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体n。
[0092][0093]
(14)制备中间体o:将化合物n溶解在(1~100ml)的二氯甲烷中,在0℃冰浴条件下依次添加nahco3(1~10g)以及戴斯-马丁试剂(1~10g),在冰浴条件下反应10分钟,使用tlc检测反应完全后,使用na2o3s2淬灭,加水稀释加etoac萃取,饱和食盐水、洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,得到无定型固体化合物,经质谱、一维和二维核磁共振鉴定化合物为中间体o。
[0094][0095]
(15)制备毛萼内酯素b:将化合物o溶解于(1~50ml)甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,得到无定型白色固体化合物,经质谱、一维和二维核磁共振鉴定化合物为毛萼内酯素b。
[0096][0097]
实施例2
[0098]
(1)制备中间体b:将化合物a(20.0g,58mmol,1.0equiv)溶解在30ml的二氯甲烷中,加入二异丙基乙胺(11.5ml,87mmol,1.5equiv)。在0℃冰浴下缓慢滴加sem-cl(15.0ml,87mmol,1.5equiv)。然后将反应置于室温中反应4个小时,tlc检测反应完全后,饱和nahco3(30ml)淬灭,etoac(3
×
300ml)萃取,饱和食盐水(50ml)洗涤,、无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=12:1,得到无定型固体化合物b。data for b:rf=0.41(silica,petroleum ether/acetone 4:1);4:1);(meoh,c 0.07);1h nmr(600mhz,cdcl3)δ=6.71(d,j=10.1,1h),6.29(s,1h),5.86(d,j=10.1,1h),5.82(s,1h),5.25(d,j=1.4,1h),5.02(d,j=7.1,1h),4.77(d,j=7.1,1h),4.34(dd,j=10.3,1.3,1h),4.07(ddd,j=12.6,9.3,4.9,1h),4.00
–
3.93(m,2h),3.54(ddd,j=12.0,9.3,5.8,1h),3.00(dd,j=9.8,4.5,1h),2.41(d,j=9.1,1h),2.34
–
2.25(m,2h),2.18(d,j=19.1,2h),2.12
–
2.02(m,1h),1.97
–
1.92(m,1h),1.40
–
1.30(m,2h),1.23(d,j=17.0,6h),0.96(td,j=13.0,5.8,1h),0.89
–
0.80(m,1h),-0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=203.1,197.3,160.8,153.5,127.5,115.2,97.6,95.4,86.1,67.3,65.4,58.4,54.7,47.9,47.1,36.1,33.9,30.4,29.6,25.9,25.6,19.2,17.9,
–
1.4;hresims(m/z):[m na]
calcd for c
26h38
o6sina
497.2330,found 497.2335.
[0099]
(2)制备中间体c:将化合物b(18.0g,38mmol,1.0equiv)溶解在20ml的四氢呋喃中,置于
–
50℃的低温反应器中充分冷却15分钟。后缓慢加入nabh4(2.8g,76mmol,2.0equiv),充分反应30分钟。tlc检测反应完全后,使用丙酮淬灭,etoac(3
×
400ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=85:15,得到无定型固体化合物c。data for c,rf=0.32(silica,petroleum ether/acetone 4:1);(meoh,c 0.04);mp 218
–
222℃,1h nmr(600mhz,cdcl3)δ=6.67(d,j=10.1,1h),5.87(d,j=10.1,1h),5.10(s,1h),5.10(s,1h),5.07(d,j=1.4,1h),4.96(d,j=6.9,1h),4.83(d,j=6.9,1h),4.38(d,j=2.7,1h),4.35(d,j=10.0,1h),4.15(d,j=2.6,1h),3.92
–
3.88(m,2h),3.85(ddd,j=11.4,9.5,5.5,1h),3.62(ddd,j=11.3,9.4,6.1,1h),2.62(dd,j=8.9,5.1,1h),2.48(d,j=7.9,1h),2.35
–
2.27(m,1h),2.01(s,1h),1.88
–
1.82(m,1h),1.80(d,j=12.2,1h),1.65(dd,j=12.3,5.1,1h),1.38(td,j=12.4,7.4,1h),1.30
–
1.24(m,1h),1.22(s,3h),1.16(s,3h),1.07
–
0.96(m,1h),0.03(s,9h);
13
c nmr(151mhz,cdcl3)δ=198.0,159.5,159.4,128.0,108.4,97.1,95.7,84.7,74.6,67.7,65.1,54.3,52.7,46.6,40.8,36.0,35.5,32.5,31.1,25.4,25.0,18.1,17.6,
–
1.4;hresims(m/z):[m na]
calcd for c
26h40
o6sina
499.2486,found 499.2475.
[0100]
(3)制备中间体d:将化合物c(10.0g,21mmol,1.0equiv)溶解在10ml的甲醇中,先添加六水合二氯化镍充分搅拌半小时;后置于
–
78℃的低温反应器中充分冷却半个小时。分三次缓慢加入nabh4(7.7g,210mmol,10.0equiv),加入后反应液立刻变黑,充分反应1小时
后,将反应移至室温继续反应1个小时。tlc检测反应完全后,饱和nahco3(30ml)淬灭,淬灭后添加40g硅藻土,搅拌均匀后分别使用etoac(3
×
400ml)抽滤,抽滤后加水萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=85:15,得到无定型固体化合物d。data for d,rf=0.38(silica,petroleum ether/acetone 4:1);(meoh,c 0.04);1h nmr(600mhz,cdcl3)δ=5.25(d,j=2.1,1h),5.14(s,1h),5.12
–
5.10(m,1h),4.91(d,j=7.0,1h),4.78(d,j=7.1,1h),4.39(d,j=2.8,1h),4.34(d,j=10.4,1h),4.16(t,j=1.8,1h),3.89
–
3.79(m,2h),3.72(d,j=7.9,1h),3.60(ddd,j=11.5,9.5,6.1,1h),2.68
–
2.59(m,2h),2.44(d,j=8.0,1h),2.39(dd,j=13.4,5.7,1h),2.24(ddd,j=14.9,9.1,2.9,1h),2.12(dd,j=13.4,8.8,1h),1.95
–
1.88(m,1h),1.72(d,j=12.3,1h),1.67(m,2h),1.64(m,2h),1.40(td,j=12.4,7.7,1h),1.06
–
1.00(m,1h),0.98(s,3h),0.96(s,3h),0.96
–
0.92(m,1h),0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=213.6,159.3,108.7,97.1,95.6,85.4,74.7,67.6,64.4,57.1,52.3,49.0,40.8,38.1,35.5,35.5,33.0,32.3,30.1,25.4,24.1,18.0,17.3,
–
1.4;hresims(m/z):[m na]
calcd for c
26h42
o6sina
501.2643,found 501.2637.
[0101]
(4)制备中间体e:将化合物d(7.0g,15mmol,1.0equiv)溶解在7ml的乙腈中,依次加入dbu(6.6ml,44mmol,3.0equiv)和对甲基苯磺酰氯叠氮(5.8ml,75%in ea,22mmol,1.5equiv)。该反应置于室温中充分反应2小时。tlc检测反应完全后,使用饱和nahco3(30ml)淬灭,etoac(3
×
300ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=6:1,得到无定型黄色固体化合物e。data for e,rf=0.38(silica,petroleum ether/acetone 3:1);(meoh,c 0.05);mp 218
–
222℃;1h nmr(600mhz,cdcl3)δ=5.17(d,j=1.5,1h),5.11
–
5.09(s,1h),5.09(s,1h),4.92(d,j=7.1,1h),4.80(d,j=6.9,1h),4.38
–
4.31(m,2h),4.10(dd,j=2.7,1.2,1h),3.89(dd,j=9.9,1.9,1h),3.87
–
3.81(m,1h),3.70(d,j=8.0,1h),3.60(ddd,j=11.8,9.3,6.2,1h),2.78(d,j=13.9,1h),2.61(dd,j=8.8,5.1,1h),2.29(dd,j=13.4,4.7,1h),2.24(d,j=13.9,1h),2.18
–
2.15(m,2h),1.88
–
1.83(m,1h),1.79(d,j=12.3,1h),1.63(dd,j=12.3,5.1,1h),1.36(dt,j=11.2,6.5,2h),1.13(s,3h),1.07(s,3h),1.04
–
0.94(m,2h),0.02(d,j=1.1,9h);
13
c nmr(151mhz,cdcl3)δ=192.5,159.6,108.4,97.2,95.4,85.2,74.7,67.7,65.1,63.9,56.0,52.7,47.1,41.4,37.9,35.4,33.6,32.6,29.2,25.4,21.3,18.1,17.8,
–
1.4;hresims(m/z):[m na]
calcd for c
26h40
n2o6sina
527.2548,found 527.2542.
[0102]
(5)制备中间体f:将化合物e(4.0g,10mmol)溶解于4ml甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=6:1,得到无定型白色固体化合物f。data for f,rf=0.30(silica,petroleum ether/acetone 4.5:1);(meoh,c 0.05);1h nmr(400mhz,acetone-d6)δ=5.13(s,1h),5.10
–
5.03(m,1h),4.83(d,j=6.9,1h),4.78(d,j=7.0,1h),4.59(dt,j=5.4,2.7,1h),4.49(d,j=1.7,2h),4.35(d,j=12.5,1h),3.84
–
3.77(m,1h),3.69
–
3.60(m,1h),2.75(d,j=5.9,1h),2.51(t,j=7.2,1h),2.26(d,j=12.6,1h),2.23
–
2.13(m,2h),2.09(s,1h),2.03(s,1h),1.89(dd,j=13.7,2.7,1h),1.78(d,j=12.6,1h),1.70
–
1.63(m,1h),1.61
–
1.50(m,2h),1.44(dt,j=13.8,
7.0,1h),1.20(s,3h),1.13(s,3h),0.99
–
0.92(m,2h),0.02(s,9h);
13
c nmr(101mhz,acetone-d6)δ=210.7,180.4,158.2,108.2,95.2,79.4,77.7,70.5,67.0,60.0,60.0,55.4,47.6,47.5,42.7,41.8,38.5,32.8,32.5,32.1,22.0,19.2,18.8,
–
1.2;hresims(m/z):[m na]
calcd for c
26h40
o6sina
499.2486,found 499.2483.
[0103]
(6)制备中间体g:将化合物f(2.9g,5.5mmol,1.0equiv)溶解在2ml的二氯甲烷中,反应置于室温中依次添加dhp(2.0ml,22mmol,4.0equiv)与对甲苯磺酸吡啶盐(305mg,1.2mmol,0.2equiv)。室温充分反应15分钟,使用tlc检测反应完全后,使用饱和nahco3(30ml)淬灭,etoac(3
×
400ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=10:1,得到无定型油状化合物g1和g2。
[0104]
data for g1:rf=0.29(silica,petroleum ether/acetone 6:1);6:1);(meoh,c 0.08);1h nmr(600mhz,cdcl3)δ=5.44
–
5.39(m,1h),5.12(d,j=2.8,1h),4.93(d,j=7.1,1h),4.79(d,j=7.1,1h),4.75
–
4.72(m,1h),4.60
–
4.57(m,1h),4.48(s,2h),4.26(d,j=13.3,1h),4.04
–
3.98(m,1h),3.79(ddd,j=11.6,9.5,5.5,1h),3.58(ddd,j=11.5,9.5,5.7,1h),3.50(ddd,j=11.6,8.3,3.3,1h),2.77(d,j=5.8,1h),2.73(dd,j=11.7,2.9,1h),2.38(t,j=7.1,1h),2.18
–
2.08(m,2h),2.03
–
1.93(m,2h),1.86
–
1.76(m,2h),1.74
–
1.66(m,2h),1.65
–
1.59(m,1h),1.56
–
1.47(m,5h),1.41(dt,j=14.1,7.0,1h),1.20(s,3h),1.13(s,3h),0.98(ddd,j=13.6,11.7,5.7,1h),0.91(ddd,j=13.5,11.5,5.4,1h),0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=211.4,180.2,154.5,110.2,101.4,95.0,84.9,76.7,70.0,66.8,64.9,58.9,58.8,55.2,47.5,46.9,42.6,41.4,37.7,32.7,32.0,31.8,31.2,25.4,21.7,21.3,18.7,18.3,
–
1.3;hresims(m/z):[m na]
calcd for c
31h48
o7sina
583.3062,found583.3067.
[0105]
data for g2:rf=0.32(silica,petroleum ether/acetone 6:1);6:1);(meoh,c 0.05);1h nmr(600mhz,cdcl3)δ=5.20
–
5.17(m,1h),5.08(d,j=2.8,1h),4.94(t,j=2.7,1h),4.90(d,j=7.5,1h),4.86(d,j=7.5,1h),4.82(d,j=10.1,1h),4.71(d,j=10.1,1h),4.45
–
4.39(m,1h),4.33(d,j=14.0,1h),3.85(ddd,j=12.0,9.5,5.3,1h),3.75
–
3.69(m,1h),3.56(ddd,j=11.8,9.5,5.7,1h),3.20(td,j=11.2,3.1,1h),2.79
–
2.74(m,2h),2.69(d,j=9.2,1h),2.25(d,j=14.0,1h),2.10(dd,j=13.8,11.9,1h),1.98(dd,j=13.8,2.3,1h),1.96
–
1.90(m,1h),1.86
–
1.78(m,2h),1.79
–
1.72(m,1h),1.64(d,j=12.3,2h),1.54
–
1.40(m,4h),1.39
–
1.31(m,2h),1.19(s,3h),1.13(s,3h),0.98(ddd,j=13.6,12.0,5.6,1h),0.90(ddd,j=13.5,11.7,5.2,1h),0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=208.5,180.8,151.9,108.6,104.2,95.7,84.7,77.4,71.3,66.6,64.9,59.5,56.7,56.5,48.2,47.3,41.4,40.3,38.6,37.1,32.2,31.9,31.5,25.0,22.3,20.4,19.1,18.2,
–
1.3;hresims(m/z):[m na]
calcd for c
31h48
o7sina
583.3062,found 583.3060.
[0106]
(7)制备中间体h:将化合物e1和e2(1.3g 1.2g,4.5mmol,1.0equiv)溶解在2ml的四氢呋喃中,反应置于
–
40℃的低温反应器中充分冷却10分钟后,先添加lda(5.0ml,2m in thf,9mmol,2.0equiv)与底物反应40分钟。后加入oxaziridine(2.3g,9mmol,2.0equiv)(cas:63160-13-40),
–
40℃继续充分反应2个小时,使用tlc检测反应完全后,使用饱和nahco3(30ml)淬灭,etoac(3
×
100ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,
浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=8:1,得到无定型油状化合物h1和h2。
[0107]
data for h1:rf=0.30(silica,petroleum ether/acetone 5:1);5:1);(meoh,c 0.07);1h nmr(600mhz,cdcl3)δ=5.39(s,1h),5.11(s,1h),4.88(d,j=7.1,1h),4.83(d,j=7.0,1h),4.77(s,1h),4.62(dd,j=6.5,2.5,1h),4.55(d,j=10.3,1h),4.50(d,j=10.2,1h),4.10(d,j=13.5,1h),4.05
–
3.98(m,1h),3.77(ddd,j=11.7,9.5,5.6,1h),3.59(ddd,j=11.6,9.5,5.8,1h),3.54
–
3.48(m,1h),2.76(s,1h),2.74
–
2.68(m,1h),2.60(s,1h),2.41(d,j=13.4,1h),2.25(d,j=15.4,1h),2.05(d,j=15.3,1h),1.91
–
1.86(m,1h),1.86
–
1.80(m,1h),1.80
–
1.75(m,1h),1.72
–
1.63(m,3h),1.58
–
1.48(m,6h),1.29(s,3h),1.26(s,3h),0.96(ddd,j=13.6,11.6,5.7,1h),0.89(ddd,j=13.8,11.5,5.5,1h),0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=211.4,179.1,154.5,110.0,100.8,95.4,84.3,80.6,77.6,69.5,66.9,64.5,58.7,58.3,57.1,56.8,39.9,38.0,35.5,34.9,32.9,31.7,31.1,25.5,25.5,20.9,18.9,18.2,
–
1.3;hresims(m/z):[m na]
calcd for c
31h48
o8sina
599.3011,found599.3011.
[0108]
data for h2:rf=0.32(silica,petroleum ether/acetone 5:1);5:1);(meoh,c 0.07);1h nmr(400mhz,cdcl3)δ=5.22
–
5.14(m,1h),5.05(d,j=2.9,1h),4.99
–
4.83(m,4h),4.72(d,j=10.2,1h),4.52(dd,j=7.7,2.0,1h),4.15(d,j=14.0,1h),3.87
–
3.73(m,2h),3.57(ddd,j=11.7,9.5,5.7,1h),3.32
–
3.19(m,1h),2.98(d,j=8.8,1h),2.89
–
2.71(m,2h),2.50(d,j=14.0,1h),2.30(d,j=15.2,1h),2.04(d,j=15.2,1h),1.97
–
1.73(m,5h),1.71
–
1.64(m,1h),1.64
–
1.52(m,2h),1.52
–
1.37(m,4h),1.24(s,3h),1.24(s,3h),0.92(dtd,j=30.7,13.4,5.6,2h),0.01(s,9h);
13
c nmr(101mhz,cdcl3)δ=208.6,179.8,152.2,108.3,103.3,96.0,84.3,81.0,78.2,70.4,66.7,64.5,59.0,58.7,57.4,55.5,39.7,38.8,37.2,34.6,33.8,31.8,31.4,25.1,24.1,21.7,19.1,18.2,
–
1.3;hresims(m/z):[m na]
calcd for c
31h48
o8sina
599.3011,found 599.3005.
[0109]
(8)制备中间体i:将化合物h1和h2(1.3g,2.6mmol,1.0equiv)溶解在1ml的乙醇中,添加对甲苯磺酸吡啶盐(113mg,0.5mmol,0.2equiv)在68℃中反应1个小时。使用饱和nahco3(30ml)淬灭,etoac(3
×
100ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=5:1,得到无定型固体化合物i。data for i:rf=0.41(silica,petroleum ether/acetone 3:1);(meoh,c 0.04);1h nmr(600mhz,cdcl3)δ=5.20(s,1h),5.15(s,1h),4.88(d,j=7.0,1h),4.69(d,j=7.1,1h),4.38(d,j=3.2,1h),4.15(d,j=10.1,1h),4.09(dd,j=14.6,10.5,2h),3.77(ddd,j=11.5,9.4,5.2,1h),3.54(ddd,j=11.6,9.5,5.7,1h),3.34(d,j=3.9,1h),2.81(dd,j=11.5,5.1,1h),2.75
–
2.72(m,2h),2.60(s,1h),2.24
–
2.14(m,2h),2.06(d,j=15.3,1h),2.01(m,1h),1.80(dd,j=12.7,5.2,1h),1.54
–
1.41(m,1h),1.30(s,3h),1.29
–
1.24(m,2h),1.20(s,3h),0.98(ddd,j=13.7,11.8,5.8,1h),0.90(ddd,j=13.9,11.7,5.4,1h),0.02(s,9h);
13
c nmr(151mhz,cdcl3)δ=210.8,178.6,156.4,109.7,93.6,80.4,80.3,77.1,68.8,66.9,58.1,57.8,57.6,56.2,39.4,36.8,36.3,34.6,32.2,29.1,26.8,18.4,18.2,
–
1.3;hresims(m/z):[m na]
calcd for c
26h40
o7sina
515.2436,found 515.2433.
[0110]
(9)制备中间体j:将化合物i(700mg,1.4mmol,1.0equiv)溶解在1ml的二氯甲烷中,在冰浴条件下依次添加乙酸酐(180μl,1.7mmol,1.2equiv),dmap(210mg,1.7mmol,
1.2equiv)中反应1个小时。使用饱和nahco3(30ml)淬灭,etoac(3
×
50ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=5:1,得到无定型固体化合物j。data for j:rf=0.34(silica,petroleum ether/acetone 4:1);(meoh,c 0.04);mp 230
–
232℃;1h nmr(600mhz,cdcl3)δ=5.68(s,1h),5.10(s,1h),4.93(dd,j=2.6,1.4,1h),4.78(dd,j=7.3,1.0,1h),4.57(dd,j=7.3,1.1,1h),4.24(d,j=10.2,1h),4.19(d,j=10.2,1h),4.07(d,j=11.8,1h),3.75(m,1h),3.54
–
3.46(m,1h),2.88(s,1h),2.85
–
2.76(m,2h),2.67
–
2.62(m,1h),2.21(d,j=15.3,1h),2.16(s,1h),2.12
–
1.98(m,3h),1.88(dd,j=12.7,5.2,1h),1.68(d,j=12.7,1h),1.54
–
1.52(m,2h),1.32(s,3h),1.30
–
1.24(m,2h),1.22(s,3h),1.01
–
0.92(m,1h),0.92
–
0.82(m,1h),0.01(s,9h);
13
c nmr(151mhz,cdcl3)δ=209.3,178.8,170.3,153.4,109.9,93.7,80.5,78.4,76.4,68.8,66.6,57.8,57.6,56.4,56.2,39.6,37.6,36.9,34.9,31.9,30.5,26.3,21.4,18.6,18.2,
–
1.3;hresims(m/z):[m na]
calcd for c
28h42
o8sina
557.2541,found 557.2539.
[0111]
(10)制备毛萼内酯素a:将化合物j(500mg,0.9mmol)溶解在1ml的四氢呋喃中,在50℃加热条件下添加三氟乙酸(300μl)反应1个小时。使用饱和nahco3(30ml)淬灭,etoac(3
×
50ml)萃取,饱和食盐水(50ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=5:1,得到无定型固体化合物毛萼内酯素a。data for maoelactone a:rf=0.31(silica,petroleum ether/acetone 2:1);(meoh,c 0.10);1h nmr(600mhz,pyridine-d5)δ=8.75(s,1h),6.40(m,1h),5.11(br d,1h),5.00(br d,1h),4.87(d,j=10.5,1h),4.75(d,j=10.5,1h),4.57(dd,j=13.5,3.5,1h),3.27(t,j=6.5,1h),2.84(d,j=13.5,1h),2.66(m,1h),2.42(d,j=15.0,1h),2.33(d,j=15.0,1h),2.06(m,1h),2.05(s,3h),1.99(m,1h),1.88(dt,j=12.9,4.2,1h),1.76(m,2h),1.50(s,3h),1.46(s,3h),1.41(m,1h);
13
c nmr(151mhz,pyridine-d5)δ=212.3,179.6,170.3,153.5,108.8,81.2,78.6,73.6,69.4,59.3,59.2,57.4,56.0,39.9,39.0,36.8,35.7,33.0,31.6,25.9,21.0,19.6;hresims(m/z):[m na]
calcd for c
22h28
o7na
427.1727,found 427.1735.
[0112]
(11)制备中间体l:将化合物k(12g,33.0mmol)溶解在50ml的丙酮中,在0℃冰浴条件下添加琼斯试剂(10ml)反应15分钟,使用tlc检测反应完全后,使用异丙醇(30ml)淬灭,加水稀释后etoac(3
×
500ml)萃取,饱和食盐水(100ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/丙酮=4:1,得到无定型固体化合物l。化合物谱图与文献中数据对比一致(j.med.chem.2013,56,5048-5058)。
[0113]
(12)制备中间体m:将化合物l(9.0g,24.9mmol,1equiv)溶解在30ml的1,2-二氯乙烷中,在0℃冰浴条件下添加nabh(oac)3(6.3g,29.8mmol,1.2equiv)后随后缓慢滴加乙酸(300μl)反应10分钟,使用tlc检测反应完全后,使用丙酮(30ml)淬灭,加水稀释后etoac(3
×
500ml)萃取,饱和食盐水(100ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/丙酮=6:1,得到无定型固体化合物m。data for m,rf=0.41(silica,petroleum ether/acetone 2:1);(meoh,c 0.09);1h nmr(600mhz,acetone-d6)δ=6.66(d,j=4.3,1h),5.26
–
5.15(m,3h),5.11(d,j=3.9,1h),4.97
–
4.90(m,1h),4.84(t,j=3.7,1h),4.59(q,j=4.4,4.0,1h),4.21(dd,j=10.1,4.3,1h),3.88
–
3.81(m,
1h),3.74(dd,j=7.4,3.7,1h),2.74
–
2.62(m,2h),2.51(dd,j=9.1,4.2,1h),2.32
–
2.24(m,1h),2.23
–
2.12(m,2h),2.00
–
1.92(m,1h),1.72
–
1.55(m,2h),1.50
–
1.40(m,1h),1.10(s,3h),0.96(s,3h),0.94
–
0.88(m,1h);
13
c nmr(151mhz,acetone-d6)δ=213.4,159.6,110.3,99.8,76.0,73.3,72.5,64.8,57.3,53.3,49.1,45.8,42.6,39.2,36.3,33.3,32.9,30.8,24.3,17.4;hresims(m/z):[m na]
calcd for c
20h28
o6na
387.1778,found 387.1783.
[0114]
(13)制备中间体n:将化合物m(6.2g,17.0mmol,1.0equiv)溶解在15ml的dcm中,在0℃冰浴条件下依次添加dipea(11.3ml,68.0mmol,4.0equiv)以及tmscl(8.6ml,68.0mmol,4.0equiv),在0℃反应30分钟后减压浓缩。浓缩后产品溶解于乙腈,后依次添加dbu(3.8ml,25.5mmol,1.5equiv)和对甲苯磺酰叠氮(7.5ml,75%in ea,25.5mmol,1.5equiv)反应2小时后滴加tbaf(17.8g,68mmol,4.0equiv)反应30分钟,使用tlc检测反应完全后,加水稀释加etoac(3
×
500ml)萃取,饱和食盐水(100ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/丙酮=6:1,得到无定型固体化合物n。data for n,rf=0.29(silica,chcl3/meoh 3.8:0.2);(meoh,c 0.13);1h nmr(600mhz,acetone-d6)δ=6.82(s,1h),5.34
–
5.25(m,1h),5.22(d,j=3.1,1h),5.17(d,j=2.5,1h),5.11
–
5.07(m,1h),4.91(t,j=2.6,1h),4.82(d,j=2.6,1h),4.66(s,1h),4.20(d,j=9.5,1h),3.87(dd,j=9.6,2.0,1h),3.71(dd,j=7.9,3.1,1h),2.84(d,j=14.0,1h),2.55
–
2.44(m,3h),2.31(dt,j=12.9,8.9,1h),1.94(d,j=7.8,1h),1.88
–
1.77(m,1h),1.41(td,j=12.5,8.0,1h),1.23(dt,j=14.1,7.0,1h),1.19(s,3h),1.12(s,3h);
13
c nmr(151mhz,acetone-d6)δ=193.3,159.8,110.0,99.5,76.0,72.8,72.5,65.5,64.4,56.0,53.7,47.1,45.5,43.3,38.0,33.6,33.1,30.5,21.2,17.9;hresims(m/z):[m na]
calcd for c
20h26
n2o6na
413.1683,found 413.1680.
[0115]
(14)制备中间体o:将化合物n(4.6g,9.4mmol,1.0equiv)溶解在10ml的dcm中,在0℃冰浴条件下依次添加nahco3(2.4g,28.2mmol,3.0equiv)以及戴斯-马丁试剂(4.0g,9.4mmol,1.0equiv),在0℃反应10分钟,使用tlc检测反应完全后,使用na2o3s2淬灭,加水稀释加etoac(3
×
500ml)萃取,饱和食盐水(100ml)洗涤,无水mgso4干燥,过滤,浓缩后柱层析纯化,洗脱剂:石油醚/丙酮=6:1,得到无定型固体化合物o。data for o,rf=0.31(silica,petroleum ether/acetone 2:1);(meoh,c 0.07);1h nmr(600mhz,acetone-d6)δ=6.68(s,1h),6.09(s,1h),5.61(s,1h),5.50(d,j=11.4,1h),5.36(s,1h),4.96(s,1h),4.20(dd,j=9.9,1.5,1h),3.94(dd,j=9.9,1.7,1h),3.68(dd,j=11.4,8.8,1h),3.00(d,j=9.5,1h),2.83(d,j=14.0,1h),2.50
–
2.44(m,2h),2.18
–
2.14(m,1h),2.07
–
2.06(m,1h),1.72(dd,j=8.8,1.4,1h),1.59
–
1.53(m,1h),1.51
–
1.46(m,1h),1.24(s,3h),1.13(s,3h);
13
c nmr(151mhz,acetone-d6)δ=207.6,192.4,153.2,120.7,99.1,73.5,73.3,65.9,64.9,62.8,59.9,51.2,47.5,44.0,38.1,34.0,31.0,30.1,21.2,20.2;hresims(m/z):[m na]
calcd for c
20h24
n2o6na
411.1527,found 411.1521.
[0116]
(15)制备毛萼内酯素b:将化合物o(3.6g,9.4mmol)溶解于4ml甲苯中,放置于110℃加热搅拌下充分反应两个小时。tlc检测反应完全后,将反应挪到室温,待冷却后,直接减压浓缩,浓缩后柱层析纯化,洗脱剂:石油醚/乙酸乙酯=6:1,得到无定型白色固体化合物毛萼内酯素b。data for maoelactone b,rf=0.30(silica,chcl3/meoh 3.8:0.2);
(meoh,c 0.20);1h nmr(600mhz,cdcl3)δ=6.26(s,1h),5.65(s,1h),4.64(s,1h),4.37(d,j=10.4,1h),4.21(t,j=10.7,2h),4.05(d,j=10.1,1h),3.83(s,1h),3.16(d,j=6.9,1h),2.66(dd,j=11.4,3.2,1h),2.40(ddd,j=14.2,8.4,5.7,2h),2.12(dd,j=14.1,11.4,1h),2.07(d,j=12.0,1h),2.03(dd,j=14.0,3.2,1h),1.82(dtd,j=14.1,7.9,1.9,1h),1.69
–
1.58(m,2h),1.23(s,3h),1.15(s,3h);
13
c nmr(151mhz,cdcl3)δ=208.9,201.4,178.9,147.6,122.5,73.9,72.9,68.9,68.8,62.9,54.0,52.5,46.8,46.4,43.7,41.3,31.6,29.8,22.1,19.3;hresims(m/z):[m na]
calcd for c
20h24
o6na
383.1465,found 383.1475.
[0117]
实施例3
[0118]
本发明化合物细胞水平抗肿瘤活性测试。
[0119]
1.实验目的
[0120]
进行本发明化合物的抗肿瘤活性测试,通过测定化合物对人源肿瘤细胞的生长抑制活性来评价化合物的体外抗肿瘤活性。
[0121]
2.实验材料
[0122]
人非小细胞肺癌细胞株a549、人肝癌细胞hepg2、人源慢性髓系白血病细胞k562和人急性早幼粒白血病细胞hl60均为中科院上海药物研究所李佳研究员课题组馈赠,人源乳腺癌细胞mda-mb-435购自中国科学院上海生命科学研究院细胞资源中心。
[0123]
3.测试原理
[0124]
用mts比色法检测细胞增殖。mts为一种全新的mtt类似物,全称为3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2h-tetrazolium,是一种黄颜色的染料。活细胞线粒体中琥珀酸脱氢酶能够代谢还原mts,生成可溶性的甲臜(formazan)化合物,甲臜的含量可以用酶标仪在490nm处进行测定。在通常情况下,甲臜生成量与活细胞数成正比,因此可根据光密度od值推测出活细胞的数目。
[0125]
4.实验方法
[0126]
(1)接种细胞:用含10%胎牛血清的培养液(dmem或者rmpi1640)配成单个细胞悬液,以每孔3000~5000个细胞接种到96孔板,每孔体积100μl,细胞提前12~24小时接种培养。
[0127]
(2)加入待测化合物溶液:化合物用dmso溶解,化合物以40μm浓度初筛,每孔终体积200μl,每种处理均设3个复孔。
[0128]
(3)显色:37摄氏度培养48小时后,细胞弃孔内培养液,每孔加mts溶液20μl和培养液100μl;设3个空白复孔(mts溶液20μl和培养液100μl的混合液),继续孵育2~4小时,使反应充分进行后测定光吸收值。
[0129]
(4)比色:选择492nm波长,多功能酶标仪(multiskan fc)读取各孔光吸收值,记录结果,数据处理后以化合物编号为横坐标,细胞抑制率为纵坐标绘制细胞的抑制率图。
[0130]
(5)阳性对照化合物:每次实验均设顺铂(ddp)和紫杉醇(taxol)两个阳性化合物,以浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线,应用两点法(reed and muench法)计算化合物的ic50值。
[0131]
5.化合物测试结果
[0132]
表1.化合物对多种肿瘤细胞的增殖抑制活性
[0133][0134]
制剂实施例
[0135]
1.取毛萼内酯素a或/和毛萼内酯素b,按其与赋形剂重量比1:1的比例加入赋形剂,制粒压片。
[0136]
2.取毛萼内酯素a或/和毛萼内酯素b,按其与赋形剂重量比1:2的比例加入赋形剂,制粒压片。
[0137]
3.取毛萼内酯素a或/和毛萼内酯素b,按常规胶囊制剂方法制成胶囊。
[0138]
4.取毛萼内酯素a或/和毛萼内酯素b,再按下述方法制成片剂:
[0139][0140]
5.胶囊剂:取毛萼内酯素a或/和毛萼内酯素b 100mg,淀粉适量,硬脂酸模适量,制备方法:将化合物与助剂混合,过筛,在合适的容器中均匀混合,把得到的混合物装入硬明胶胶囊。
[0141]
6.鼻喷雾剂:取毛萼内酯素a或/和毛萼内酯素b 80mg
[0142][0143]
制备方法:搅拌下于适当体积的重蒸榴水中每次加入一种成分,直至完全溶解,然后再加入另一种成分。加水至2ml后,将该溶液在无菌过滤器上过滤,装入瓶中并按照适当的剂量分隔。
[0144]
7.滴丸:取毛萼内酯素a或/和毛萼内酯素b1g,聚乙二醇60009g。制法:按上述处方量称取毛萼内酯素a,加入适量无水乙醇,微热溶解后,加入处方量的聚乙二醇熔融液中(60℃水浴保温),搅拌混合均匀,直至乙醇挥尽为止,静置于60℃水浴中保温30分钟,待气泡除尽,将除尽气泡的上述混匀熔融液转入贮液筒内,在80~85℃保温条件下,控制滴速,逐滴滴入冷凝液中,待冷凝完全,倾去冷凝液,收集滴丸,沥净和用滤纸除去丸上的冷凝液,放置硅胶干燥器中或自然干燥即可。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。