一种基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法
技术领域
1.本发明属于植物分子鉴定技术领域,具体涉及一种基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法,一种联合核dna片段its核苷酸序列和叶绿体dna片段matk核苷酸序列对睡莲杂种(nymphaea hybrid——自然或人工杂交,参见2016年国际栽培植物命名法规第九版chapter i article 1)进行分子鉴定的方法。
背景技术:
2.睡莲是睡莲科睡莲属(nymphaea)植物的泛称,全世界分布广泛,睡莲属约有50种,我国产5种。睡莲属可分为5个亚属,按生态生态适应性也可划分为耐寒睡莲类和热带睡莲类。睡莲品种繁多,花色丰富,被誉为“池塘调色板”,有长达4000年的栽植历史。睡莲的拉丁属名的词根nymph为希腊神话中的山林水泽的精灵仙女。睡莲全世界约有2000个品种(http://www.victoria-adventure.org/waterlilies/names/names_a_z.htm)。睡莲花色丰富,花期长,适应力和抗逆性强,容易栽培,既有各种花色的观花品种,也有色彩斑斓的观叶品种,还有芳香宜人的闻香品种。睡莲是埃及、印度、孟加拉和斯里兰卡等国的国花,有着悠久的利用历史。除其重要的观赏价值外,睡莲也有重要的文化和经济价值,睡莲的叶片、叶柄、花梗和花是热带常食蔬菜,块茎的淀粉也可食用,同时睡莲富含多种多酚和黄酮类化合物,有一定的药用价值(selvakumari等,2016)。在生态上,睡莲亦可用于净化水体(ziarati等,2015)。睡莲的新品种繁育、食用保健等相关的产业在国内外已经初具规模(李淑娟等,2019)。睡莲属栽培广泛,但是不少杂交品种缺少完整的杂交记录信息,不清楚其父母本,对品种鉴定和进一步杂交选育提出了挑战。
3.分子鉴定不同于形态鉴定,可以从物种的dna序列信息得到形态上难以得到的特异性特征,从而用以鉴定形态上难以区分鉴别的物种和品种。叶绿体基因组在被子植物中绝大多数是单亲遗传且是母系遗传,而核基因则为双亲遗传(o'kane等,1996)。核基因组中的its序列是核糖体dna(rdna)中的内转录间隔区(internal transcribed spacer)片段。真核生物的核糖体由核糖体rna(rrna)和核糖体蛋白构成,rrna由rdna编码。植物中18s、5.8s和25s(s为沉降系数)的rrna由45s的rdna转录单位经聚合酶i辅助转录。植物中its序列共有两段,its1和its2(长度均为240bp左右),its1位于18s rrna和5.8s rrna之间,its2位于5.8s rrna和25s rrna之间。杂交起源的物种很多保留了双亲的its序列的变异位点。叶绿体基因组中的matk基因进化速率快、易扩增也已被广泛用于植物条形码技术鉴定。
4.目前,现有技术中未见有基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法的报道。
技术实现要素:
5.本发明的目的在于提供一种基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法。本发明要解决的问题在于现有技术中缺乏有效的睡莲杂交种两个亲本的鉴定方法。
6.为了实现本发明的上述目的,本发明提供了如下的技术方案:
7.一种基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法,该方法包括下述步骤:构建睡莲属its和matk核酸序列数据库,然后设计睡莲属适用引物进行pcr扩增,最后将单克隆测序的its和matk序列与已建立的对应的序列数据库比对,依据遗传距离构建邻接树并检查特异位点,得到睡莲杂种父本和母本的物种信息。
8.一种基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法,该方法包括下述步骤:
9.构建睡莲属its和matk核苷酸序列数据库:下载已有的睡莲属its和matk核苷酸序列;采取严格的过滤步骤,对its和matk序列每个“种单元”物种/亚种/变种/变型/杂交种保留一条序列;最后,比对后调整序列方向不一致的序列,然后重新比对后分别得到睡莲属its和matk核酸序列的数据库;整理最终得到的序列的序列名,格式为“物种名/序列的accession”;
10.制备睡莲属待测定亲本的物种的its单克隆序列和设计引物测序的matk核苷酸序列:提取待测定亲本的睡莲的基因组dna;设计引物;pcr序列扩增;一代测序;序列比对、构建邻接树,最后将单克隆测序的its和matk序列与已建立的对应的序列数据库比对,依据遗传距离构建邻接树并检查特异位点,得到睡莲杂种父本和母本的物种信息。
11.根据所述的基于its序列和matk序列鉴定睡莲杂种的两个亲本的方法,其中构建睡莲属its和matk核酸序列数据库的步骤如下:
12.a.以“((nymphaea[organism])and 5.8s[title])not predicted[title]”和“((nymphaea[organism])and matk[title])not predicted[title]”语法搜索美国国家生物技术信息中心(ncbi)核酸数据库(www.ncbi.nlm.nih.gov/nucleotide),得到睡莲属全部的its和matk序列数据;同时以“(chloroplast,complete genome[title]or plastid,complete genome[title])and nymphaea[organism]”语法得到59个已发表的睡莲属叶绿体基因组,并提取其中的matk序列;
[0013]
b.采取严格的过滤步骤,对its和matk序列每个“种单元”即物种/亚种/变种/变型/杂交种保留一条序列;
[0014]
c.比对后调整序列方向不一致的序列,然后重新比对后分别得到睡莲属its和matk核酸序列的数据库。
[0015]
4.根据权利要求2所述的一种构建睡莲属its和matk核苷酸序列数据库的方法,其特征在于:所述的过滤步骤为:
[0016]
a.去掉物种名含unverified、sp.和cf.的,保留种下等级亚种、变种、变型和明确的杂交种,简称其为“种单元”,将isolate、voucher、genotype、strain等都视为栽培种,将归在每个“种单元”内部;
[0017]
b.若“种单元”仅含一条序列,则不过滤;过滤后每个“种单元”最终仅保留一条序列;
[0018]
c.若步骤a中的栽培种的序列与“种单元”的序列差异较大,则优先去掉栽培种的序列;
[0019]
d.优先去掉差异明显过大,特别是编码区如5.8s rrna的序列,优先去掉含未知碱基、简并碱基的序列,优先保留更长的序列;
[0020]
e.叶绿体基因组中提取得到matk序列仅作为补充使用,当测序得到的matk序列已有“种单元”,则优先舍弃从叶绿体基因组中提取得到的matk序列;
[0021]
f.最终保留的序列为最能代表该“种单元”的序列,即与多条序列得到的一致性序列最接近的序列。
[0022]
所述的制备睡莲属待测定亲本的物种的its序列和设计引物测序的matk序列步骤为:
[0023]
a.采用睡莲属18s、5.8s和25s rrna的保守核苷酸序列参考植物条形码its通用引物设计睡莲属适用的its引物,采用睡莲属matk的保守核苷酸序列参考植物通用条形码引物设计睡莲属适用的matk引物;
[0024]
b.提取待测定亲本的睡莲的基因组dna;
[0025]
c.以步骤b提取的待测定亲本的睡莲的基因组dna为模版,利用步骤a设计的引物进行pcr扩增,分别得到its和matk序列的扩增产物;
[0026]
d.以步骤c得到的its序列的扩增产物,使用plb零背景快速克隆试剂盒连接,并转化到dh5α感受态细胞,然后将挑取的菌斑经载体引物pcr扩增得到its序列克隆扩增的pcr扩增产物;
[0027]
e.将步骤c得到的matk序列的扩增产物和步骤d得到的its序列克隆扩增产物经一代测序仪测序,将测序结果进行序列拼接,得到待测定亲本的睡莲的matk序列和单克隆的its序列。
[0028]
所述的引物为:
[0029]
its-5,其核苷酸序列为agtcgtaacaaggtttccgt;
[0030]
its-3,其核苷酸序列为tagtaacggcgagcgaacc;
[0031]
matk-5,其核苷酸序列为cgtaccgtacttttatgtttacgag;
[0032]
matk-3,其核苷酸序列为acccaatccatctggaaatcttgcttc。
[0033]
所述pcr扩增的反应体系为:
[0034]
its序列的pcr的扩增体系为30μl,体系包含pcr 2
×
mix 15μl,ddh2o 13μl,引物各0.5μl,总dna 2.0μl。its序列的pcr扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸70s,进行35个循环;72℃延伸7min。matk序列的pcr的扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 8μl,引物各0.5μl,总dna 1.0μl。matk序列的pcr扩增条件为:95℃预变性3min;94℃变性30s,52℃退火30s,72℃延伸1min,进行35个循环;72℃延伸5min。
[0035]
所述plb连接its克隆产物pcr扩增的反应体系为:扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 10μl,引物各0.3μl,plb连接菌斑1丛。扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸80s,进行35个循环;72℃延伸7min。
[0036]
所述测序反应体系为:its和matk的测序反应的反应体系和反应条件一样。反应体系为6μl,包含bigdye 0.3μl,seqbuffer 0.5μl,ddh2o 3.75μl,单端引物0.5μl,pcr纯化产物0.5μl,反应条件为:94℃预变性30s;96℃变性10s,50℃退火5s,60℃延伸3min,进行32个循环。
[0037]
本发明的具体方法可以概括如下:
[0038]
本发明的构建睡莲属its和matk核酸序列数据库的方法,首先以“((nymphaea
[organism])and 5.8s[title])not predicted[title]”和“((nymphaea[organism])and matk[title])not predicted[title]”语法搜索美国国家生物技术信息中心(ncbi)核酸数据库(www.ncbi.nlm.nih.gov/nucleotide),得到睡莲属全部的its和matk序列数据;同时以“(chloroplast,complete genome[title]or plastid,complete genome[title])and nymphaea[organism]”语法得到59个已发表的睡莲属叶绿体基因组,并提取其中的matk序列。其次,采取严格的过滤步骤,对its和matk序列每个“种单元”(物种/亚种/变种/变型/杂交种)保留一条序列。最后,比对后调整序列方向不一致的序列,然后重新比对后分别得到睡莲属its和matk核酸序列的数据库。
[0039]
为了得到待测睡莲的its和matk序列,首先提取待测定亲本的睡莲的基因组dna,然后采用睡莲属18s、5.8s和25s rrna的保守核苷酸序列参考植物条形码its通用引物设计睡莲属适用的its引物,即引物对为正向its-5(gtcgtaacaaggtttccgt)、反向its-3(tagtaacggcgagcgaacc)。采用睡莲属matk的保守核苷酸序列参考植物通用条形码引物设计睡莲属适用的matk引物,即引物对为正向matk-5(cgtaccgtacttttatgtttacgag)、反向matk-3(acccaatccatctggaaatcttgcttc)。使用引物对its-5/its-3得到its的pcr扩增产物,使用引物对matk-5/matk-3得到matk的pcr扩增产物。its序列的pcr的扩增体系为30μl,体系包含pcr 2
×
mix 15μl,ddh2o 13μl,引物各0.5μl,总dna 2.0μl。its序列的pcr扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸70s,进行35个循环;72℃延伸7min。matk序列的pcr的扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 8μl,引物各0.5μl,总dna 1.0μl。matk序列的pcr扩增条件为:95℃预变性3min;94℃变性30s,52℃退火30s,72℃延伸1min,进行35个循环;72℃延伸5min。
[0040]
使用引物对its-5/its-3得到its的pcr扩增产物后,再使用plb零背景快速克隆试剂盒连接,并转化到大肠杆菌dh5α感受态细胞,然后将挑取的菌斑再经载体引物pcr扩增得到its序列单克隆扩增的pcr扩增产物。扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 10μl,引物各0.3μl,plb连接菌斑1丛。扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸80s,进行35个循环;72℃延伸7min。
[0041]
上机测序前需进行测序反应,its和matk的反应体系和反应条件一样。反应体系为6μl,包含bigdye 0.3μl,seqbuffer 0.5μl,ddh2o 3.75μl,单端引物0.5μl,pcr纯化产物0.5μl。反应条件为:94℃预变性30s;96℃变性10s,50℃退火5s,60℃延伸3min,进行32个循环。经一代测序仪测序,将测序结果进行序列拼接,得到待测定亲本的睡莲的matk序列和单克隆的its序列。
[0042]
得到待测物种的its和matk序列后,将其与构建的睡莲属标准数据库进行序列比对,基于遗传距离构建邻接树,然后将与待测物种聚为一枝的物种序列挑出,修剪整齐后重新比对建树,并对变异位点进行核对,从而准确确定与待测物种最接近的物种,将其定为其可能的亲本物种。
[0043]
与现有技术相比,本发明具备如下的优益性:
[0044]
本发明基于睡莲属已发表的大量分子序列数据,选择包含双亲信息的its序列和仅包含母本信息的matk序列,经过过滤去重、比对整理构建睡莲属的its和matk核酸序列数据库。然后设计睡莲属适用引物进行pcr扩增,最后将单克隆测序的its和测序的matk序列与已建立的对应的序列数据库比对,基于遗传距离构建邻接树并检查特异位点,从而得到
睡莲杂种父本和母本的物种信息。本发明的方法不仅适用于人工杂交的栽培品种,也适用于自然产生的杂交物种(符合国际植物命名法规规定的或符合国际栽培植物命名法规的)。本发明通过联合核基因组的its序列(携带两个亲本的信息)和叶绿体基因组中matk序列(携带母本信息),通过对序列比对后的特异特征以及利用遗传距离构建nj树,从而得到睡莲杂交种的父本和母本物种信息。本发明可广泛用于睡莲栽培品种的鉴定,以及睡莲育种时亲本材料的辅助验证。
附图说明
[0045]
为了更清楚地说明本发明具体实施方式,下面将对具体实施方式所需要使用的附图作简单介绍。
[0046]
图1为本发明实施例3中10条黄乔伊睡莲的单克隆测序的its序列与50条睡莲属its数据库比对并构建邻接树的结果,实线框内为与黄乔伊睡莲10条its序列聚在一起的一个分支。
[0047]
图2为本发明实施例3中从图1挑出分支后再比对构建邻接树并展示局部序列比对图,虚线框突出显示了墨西哥睡莲和球根香睡莲,实线框展示分子鉴定关键位点。
[0048]
图3为本发明实施例3中全部its比对结果,红色实线框展示用以鉴定的关键比对位点。
[0049]
图4为本发明实施例3中2条黄乔伊睡莲的matk序列与32条睡莲属matk数据库比对并构建邻接树的结果,实线框内为与黄乔伊睡莲2条matk序列聚在一起的一个分支。
[0050]
图5为本发明实施例3中从图4挑出分支后比对建树并展示局部序列比对图,实线框突出显示了matk的特异位点。
具体实施方式
[0051]
以下通过具体实施例来说明本发明的实施方式,除非另外说明,本发明中所公开的实验方法均采用本技术领域常规技术,实施例中所用到的试剂和原料均可由市场购得。为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。
[0052]
实施例1
[0053]
本实施例提供了一种构建睡莲属its和matk核苷酸序列数据库的方法,其方法可在添加网上公共数据库或新测序的序列后更新已构建的核苷酸序列数据库。具体包括以下步骤:
[0054]
1.下载已有的睡莲属its和matk核苷酸序列。以“((nymphaea[organism])and 5.8s[title])not predicted[title]”和“((nymphaea[organism])and matk[title])not predicted[title]”语法搜索美国国家生物技术信息中心(ncbi)核酸数据库(www.ncbi.nlm.nih.gov/nucleotide),得到睡莲属全部的its和matk序列数据。结果得到its序列共499条,序列长度范围为213bp~1002bp。得到matk序列共49条,去掉两个unverified的序列(kj747597和jq024977)后剩余47条,47条matk序列长度范围为556bp~2585bp。为了得到更全的物种的matk序列,另外以“(chloroplast,complete genome[title]or plastid,complete genome[title])and nymphaea[organism]”语法搜索得到
59个已发表的睡莲属叶绿体基因组,去掉鉴定不明确的物种,剩下的物种仅取refseq数据库中的,即24个“nc_”开头的物种。提取24个睡莲物种的叶绿体基因组的matk序列。
[0055]
2.采取严格的过滤步骤,对its和matk序列每个“种单元”(物种/亚种/变种/变型/杂交种)保留一条序列。最后,比对后调整序列方向不一致的序列,然后重新比对后分别得到睡莲属its和matk核酸序列的数据库。其中采取的严格过滤步骤包括:
[0056]
a.去掉物种名含unverified、sp.和cf.的,保留种下等级亚种(subsp.)、变种(var.)、变型(f.)和明确的杂交种(
×
),简称其为“种单元”。将isolate、voucher、genotype、strain等都视为栽培种(cultivar),将归在每个“种单元”内部;
[0057]
b.若“种单元”仅含一条序列,则不过滤;过滤后每个“种单元”最终仅保留一条序列;
[0058]
c.若步骤a中的栽培种的序列与“种单元”的序列差异较大,则优先去掉栽培种的序列;
[0059]
d.优先去掉差异明显过大(特别是编码区如5.8s rrna)的序列,优先去掉含未知碱基、简并碱基的序列,优先保留更长的序列;
[0060]
e.叶绿体基因组中提取得到matk序列仅作为补充使用,当测序得到的matk序列已有“种单元”,则优先舍弃从叶绿体基因组中提取得到的matk序列;
[0061]
f.最终保留的序列为最能代表该“种单元”的序列(与多条序列得到的一致性序列最接近的序列)。
[0062]
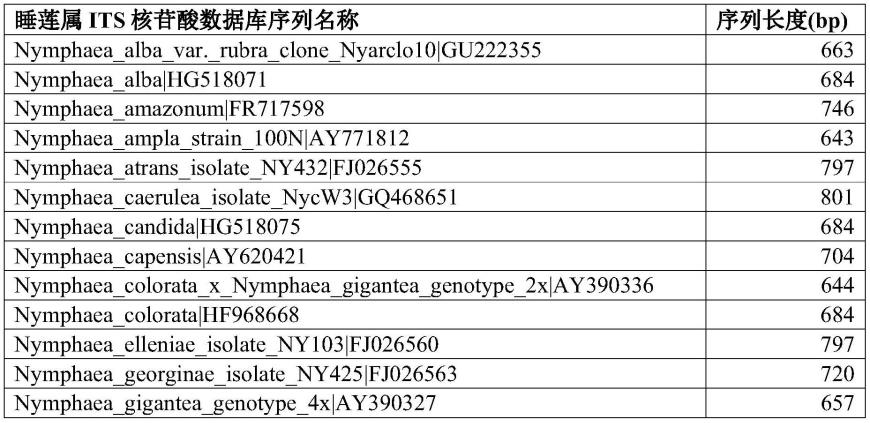
3.整理最终得到的序列的序列名,格式为“物种名|序列的accession”。最终得到的睡莲属its核苷酸数据库共50条序列(详见表1),matk核苷酸数据库共32条序列(详见表2),来源于叶绿体基因组中的序列标注有“_plastome”。
[0063]
表1睡莲属its核苷酸数据库序列信息
[0064]
[0065][0066]
表2睡莲属matk核苷酸数据库序列信息
[0067]
[0068][0069]
实施例2
[0070]
本实施例提供了一种获取睡莲属待测定亲本的物种的its(单克隆)和matk核苷酸序列的方法,具体包括以下步骤:
[0071]
1.提取待测定亲本的睡莲的基因组dna。利用改良的2
×
ctab法(rogers和bendich,1985)提取睡莲叶片的总dna。大致流程包括液氮下研磨、ctab提取液65℃水浴、氯仿异戊醇抽提、异戊醇沉降、乙醇洗涤、核糖核酸酶消化等。
[0072]
2.设计引物。下载ncbi核苷酸数据库中睡莲属的18s和25s序列数据,结合实施例1中下载的its序列数据,将植物中常用的its引物对与其搜索,找到产物大小合适、与睡莲属18s和25s序列互补性高的对应引物作为最终pcr扩增睡莲属its的引物对。最终选取的its的引物对为:正向its-5(gtcgtaacaaggtttccgt)、反向its-3(tagtaacggcgagcgaacc)。同样的,将植物中常用的matk引物对与实施例1中下载的matk序列进行搜索比对,修改不一致的碱基使其与睡莲属的matk序列互补性高。最终选取的matk的引物对为:正向matk-5(cgtaccgtacttttatgtttacgag)、反向matk-3(acccaatccatctggaaatcttgcttc)。
[0073]
3.pcr序列扩增。使用引物对its-5/its-3得到its的pcr扩增产物,使用引物对matk-5/matk-3得到matk的pcr扩增产物。its序列的pcr的扩增体系为30μl,体系包含pcr 2
×
mix 15μl,ddh2o 13μl,引物各0.5μl,总dna 2.0μl。its序列的pcr扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸70s,进行35个循环;72℃延伸7min。matk序列的pcr的扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 8μl,引物各0.5μl,总dna 1.0μl。matk序列的pcr扩增条件为:95℃预变性3min;94℃变性30s,52℃退火30s,72℃延伸1min,进行35个循环;72℃延伸5min。pcr扩增产物经1%琼脂糖凝胶电泳检测(4s green染料,150v,25min电泳),扩增产物长度应分别为900bp(its)和1000bp(matk)左右。然后用sanprep柱式pcr产物纯化试剂盒纯化pcr产物,跑琼脂糖凝胶电泳检测pcr产物是否还在未被洗脱掉。使用引物对its-5/its-3得到its的pcr扩增产物后,再使用plb零背景快速克隆试剂盒(天根ct205)用5μl体系连接粘性末端。随后将5μl连接产物加入到dh5α感受态细胞(天根cb101)中,转化后涂匀在固体lb培养基(含氨苄青霉素)上,37℃培养15小时。用枪头挑斑,洗入到载体引物pcr扩增体系中。扩增体系为20μl,体系包含pcr 2
×
mix 10μl,ddh2o 10μl,引物各0.3μl,plb连接菌斑1丛。扩增条件为:95℃预变性3min;94℃变性55s,55℃退火55s,72℃延伸80s,进行35个循环;72℃延伸7min。载体引物pcr扩增产物使用同上的1%琼脂糖凝胶电泳检测。
[0074]
4.一代测序。上机测序前需进行pcr产物纯化和测序反应,产物纯化使用纯化酶(5μl pcr产物:2μl pcr纯化酶e),37℃和80℃各15分钟。测序反应its和matk的反应体系和反应条件一样。反应体系为6μl,包含bigdye 0.3μl,seqbuffer 0.5μl,ddh2o 3.75μl,单端引物0.5μl,pcr纯化产物0.5μl。反应条件为:94℃预变性30s;96℃变性10s,50℃退火5s,60℃延伸3min,进行32个循环。测序反应的产物再经沉降剂(95%乙醇:醋酸钠20:1)沉降、75%乙醇洗脱得到纯化的测序反应产物,变性后在3730xl测序仪测序,使用geneious软件将测序结果进行序列拼接,得到待测定亲本的睡莲的matk序列和单克隆的its序列。
[0075]
5.序列比对、构建邻接树。得到待测物种的its和matk序列后,将其与构建的睡莲属标准数据库进行序列比对,基于遗传距离构建邻接树(比对和建树均使用geneious软件)。然后将与待测物种聚为一枝的物种序列挑出,修剪整齐后重新比对建树,并对变异位点进行核对,从而准确确定与待测物种最接近的物种,将其定为其可能的亲本物种。
[0076]
实施例3
[0077]
以中国科学院昆明植物研究所植物园定植的一种睡莲品种——黄乔伊(n.'joey tomocik')的叶片为材料,采用实施例2中的方法进行dna提取、pcr扩增和测序,得到其单克隆的its序列共10种“基因型”序列共10条,序列长度为855bp~935bp(参见图1、图2、图3),得到1种matk序列2条,序列长度为977bp和1008bp(参见图4、图5)。
[0078]
将黄乔伊睡莲单克隆的its序列(10条)与实施例1构建的睡莲属its核苷酸序列数据库比对、构建邻接树,结果显示,黄乔伊的10条its序列与墨西哥睡莲(n.mexican)和香睡莲(n.odorata,包含其两个变种subsp.odorata和subsp.tuberosa)聚为一个分支(clade)(参见图1)。将该分支序列提取后再比对建邻接树,树的拓扑结构未变,4条与墨西哥睡莲聚在一起,另外6条与香睡莲聚在一起,改变邻接树视图为序列视图后可以看到,its115、its180、its198的特异位点多来自于墨西哥睡莲,its88、its212、its142、its28、its213、its40的特异位点多来自于香睡莲,而且是变种球根香睡莲(n.odorata subsp.tuberosa),
因为香睡莲种和原变种都有黄乔伊睡莲种没有的位点,而its258则两者参半(参见图2)。仔细比对所有的序列可以发现,黄乔伊睡莲中测序得到的10条单克隆its序列的特异位点与墨西哥睡莲和香睡莲的位点互有交叉(参见图3)。由此说明,黄乔伊睡莲睡莲的两个亲本应为墨西哥睡莲(n.mexican)和球根香睡莲(n.odorata subsp.tuberosa),但哪个为父本哪个为母本不能确定。
[0079]
将黄乔伊睡莲的matk序列(2条)与实施例1构建的睡莲属matk核苷酸序列数据库比对、构建邻接树。结果显示,黄乔伊的matk序列与香睡莲、墨西哥睡莲、子午莲(n.tetragona)、白睡莲(n.alba)和一个杂交种(n.
×
marliacea)聚为一个分支(参见图4)。将该分支序列提取后再比对建邻接树,结果显示,黄乔伊睡莲的2条matk序列与香睡莲聚为一支,改变邻接树视图为序列视图后可以看到,黄乔伊睡莲的几处特异位点与香睡莲完全一致,而与其他三个则不一致。由此说明,黄乔伊睡莲的母本应为香睡莲(n.odorata)。结合前面its序列的鉴定结果,可以得出鉴定结论,黄乔伊睡莲的杂交亲本分别为:父本为墨西哥睡莲(n.mexican)、母本为香睡莲(n.odorata)。至于母本是否应为香睡莲的变种球根香睡莲(n.odorata subsp.tuberosa),需要睡莲属matk核苷酸数据库更新到有了两个香睡莲变种后方可最终确定。本实施例鉴定的结果与文献中对黄乔伊睡莲模糊记录的亲本一致(powell,2009)——“parented by n.mexicana on an odorata rhizome”,也从侧面验证了本发明的准确性。
[0080]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。