用于氢化的不含铬的水稳定和酸稳定的催化剂
1.本发明涉及用于氢化有机化合物中的羰基基团的改进的不含铬的cu-al催化剂,其特征在于,该催化剂以0.5至30.0重量%的量包含锆的份额。本发明另外还涉及催化剂的制备以及其在氢化有机化合物中的羰基的用途。
2.用于氢化有机化合物如酯、二酯、醛或酮中的羰基基团的催化方法在工业中具有重要意义。它们尤其用于将羧酸或其酯,特别是脂肪酸的酯转化成相应的醇。
3.在此适合作为催化剂的是基于铜与其他过渡金属元素的组合的体系。催化剂通常以粉末形式或作为成型体,特别是片料、挤出物或粒料存在。
4.wo 2004/085356描述了一种用于氢化羰基化合物的催化剂的制备,该催化剂除了铜和铝之外还包含镧、钨、钼、钛或锆的至少一种氧化物,并且另外混有铜粉末或薄片、水泥粉或石墨。
5.de 40 21 230 a1描述了一种制备醇的方法,该方法通过在由铜、锆和氧组成的铜-锆催化剂存在下氢化有机羧酸酯化合物,以获得相应的醇如高级醇或二元醇。
6.ep 0 434 062 a1的主题是一种将物质混合物氢化成相应醇的方法,其中使用了一种通过共沉淀选自mg、zn、ti、zr、sn、ni的金属、c及其混合物制备的催化剂。
7.ep 0 552 463 a1中公开的用于氢化有机化合物中的羰基基团的催化剂在其氧化物形式中具有cuaalbzrcmndod的组成,其中a》o;b》o;c》o;d》o;a》b/2;b》a/4;a》c;a》d;和x是为了保持电中性的每单位分子式所需的氧离子数。
8.一些加氢过程的起始混合物通常具有痕量的酸性化合物。这些例如是在酯化反应中作为副产物存在的羧酸。这些化合物在氢化反应的反应条件下会侵蚀催化剂并且导致机械稳定性降低,并且有时观察到催化活性金属的部分浸出,这些金属与产物流一起从反应器中排出,并且必须从中分离。此外,随着催化活性金属的逐渐释出,催化剂的催化活性也降低。
9.对于这样的反应,使用含铜和含铬的催化剂。它们通常对酸的作用具有提高的稳定性。由于更严格的环境义务,含铬催化剂的使用与越来越高的要求相关,从而需要用具有相当的催化和物理性质的环境相容的替代物替换现有的cucr体系。
10.因此在wo2011115695a1中,将含cucr的催化剂用于将醛氢化成相应的醇。
11.因此,本发明的任务在于,提供用于氢化有机化合物中的羰基基团的催化剂,该催化剂对酸性化合物或水的影响较不敏感,并且当以催化剂成型体的形式存在时,其特征还在于改进的机械稳定性。此外,该催化剂应当尤其能够用于在酸性和/或含水介质中发生的氢化。
12.该任务通过根据本发明的催化剂得以解决。
13.本发明涉及不含铬的cu-al催化剂,其特征在于,其具有在0.5至30重量%范围内的锆份额。
14.催化剂可以以各种形式存在,例如以粉末的形式或以成型体,如挤出物、球、粒料或片料的形式存在。在优选的实施方案中,催化剂成型体以片料的形式存在。
15.当催化剂成型体以片料形式存在时,片料的直径在这种情况下通常为介于2与6mm
之间,优选介于3与5mm之间。特别优选地,直径为介于4.4与4.6mm之间。片料的高度可以为介于2与6mm之间,并且优选介于2与4mm之间。特别优选地,高度为介于2.5与3.5mm之间。
16.根据本发明的催化剂成型体具有80至500n,优选150至250n,特别优选170至230n的侧压强度。优选地,根据本发明的催化剂成型体具有在3至5mm范围内的直径,在2至4mm范围内的高度和在170至230n围内的侧压强度。
17.根据本发明的催化剂成型体的借助汞孔隙测定法测量的孔体积为介于100与500mm3/g之间,优选介于150与400mm3/g之间,特别优选介于200与400mm3/g之间。
18.在根据本发明的催化剂中的在下文给出的铜、铝和锆的量涉及催化剂的氧化、非还原形式,其中元素典型地以氧化形式以cu(ii)、al(iii)和zr(iv)存在。
19.在优选的实施方案中,催化剂以其氧化形式包含在20至50重量%范围内,优选在25至40重量%范围内的量的cu,在8至29重量%范围内,优选在15至25重量%范围内的量的al,和在0.5至30重量%范围内,优选在5至20重量%范围内,特别优选在10至20重量%范围内的量的zr,基于烧失(loss on ignition)之后的催化剂的总重量计。
20.在一个实施方案中,催化剂不含有氧化或金属形式的锰。
21.根据本发明的催化剂的特征优选在于,其具有立方二氧化锆相。在一个实施方案中,除了立方二氧化锆相之外,还存在至少一种选自斜方和单斜二氧化锆的另外的zro2变体。
22.通过下述根据本发明的步骤制备根据本发明的催化剂:
23.a)合并(i)铜化合物、锆化合物和任选的另外的过渡金属化合物的至少一种水溶液a和(ii)至少一种碱性水溶液b,以形成沉淀物,其中溶液a和/或溶液b另外包括溶解的铝化合物,
24.b)分离沉淀物,任选洗涤沉淀物,
25.c)干燥沉淀物,以获得经干燥的沉淀物,
26.d)在介于200于800℃之间的温度将根据步骤c)的经干燥的沉淀物煅烧介于30min与4h之间的时长。
27.作为在步骤a)中使用的铜、铝、锆和任选的过渡金属的化合物的起始化合物,在此原则上适合的是在水、碱性或酸性水溶液中可溶的所有化合物。优选使用碳酸盐、硝酸盐、卤化物、氧化物、硫酸盐、乙酸盐或甲酸盐。
28.铝化合物在此可以已经存在于含铜和含锆的溶液a中,或者可以与沉淀剂一起以碱性水溶液b的形式添加。
29.在此如此选择步骤a)中的溶液a中的锆化合物的份额,使得最终催化剂中的锆的份额在0.5重量至30重量%范围内,基于烧失之后的催化剂的总重量计。
30.铜化合物、锆化合物和任选的另外的过渡金属化合物的至少一种水溶液a可以以铜化合物、锆化合物和任选的另外的过渡金属化合物的多种单独的水溶液的形式提供,其中这些溶液中的每种都可以包含铝化合物。例如可以提供铜化合物的一种或更多种水溶液、锆化合物的一种或更多种水溶液、铝化合物的一种或更多种水溶液,和任选的另外的过渡金属化合物的一种或更多种水溶液。或者为此也可以提供一种或更多种共同的水溶液。它们可以通过将铜化合物和/或铝化合物和/或锆化合物和/或任选的另外的过渡金属化合物溶解在共同的容器中来制备。也可以将上述单独的溶液合并成共同的溶液。
31.在一个实施方案中,将铜化合物、锆化合物和任选的另外的过渡金属化合物的至少一种水溶液a在与碱性水溶液b合并之前,加热至高于20℃的温度,例如在50℃至90℃范围内的温度,尤其是加热至约80℃,并且在此优选进行搅拌。
32.在另一实施方案中,将碱性水溶液b在合并之前,加热高于20℃的温度,例如在50℃至90℃范围内的温度,尤其是加热至约80℃,并且在此优选进行搅拌。
33.在另一实施方案中,将铜化合物、锆化合物和任选的另外的过渡金属化合物的至少一种水溶液a以及碱性水溶液b加热至在50℃至90℃范围内的温度,尤其是加热至约85℃,并且在此优选进行搅拌。
34.在一个实施方案中,步骤a)中的沉淀物的形成通过如下进行:将含有沉淀剂的碱性水溶液b引入含有溶解的铜、铝、锆和任选的过渡金属的化合物的溶液a,优选持续搅拌含金属的溶液。
35.在另一实施方案中,步骤a)中的沉淀物的形成通过如下进行:将含有沉淀剂和铝化合物的碱性水溶液b引入含有溶解的铜、锆和任选的过渡金属的化合物的溶液a,优选持续搅拌含金属的溶液。
36.在另一实施方案中,将含有沉淀剂的碱性水溶液b与含金属的溶液a一起引入共同的沉淀容器中,其中溶液a和/或溶液b另外包含溶解的铝化合物。
37.步骤a)中的合并的溶液的温度通常在10至90℃范围内,优选介于30与90℃之间,更优选在50至85℃范围内。
38.在步骤a)中的含金属化合物的沉淀期间的ph在6.0至8.0的范围内,优选在6.5至7.5的范围内,更优选在6.5至7.0的范围内。
39.在沉淀之后,分离出所产生的沉淀物。这典型地借助过滤来进行。或者,也可以通过倾析或离心来分离出沉淀物。
40.然后可以任选地使经分离的沉淀物经受一个或更多个洗涤步骤以除去任何粘附的杂质,例如过量的氢氧根离子或碱金属离子。在此,沉淀物可以直接以滤饼的形式保留在过滤腔中并且被洗涤介质,优选去离子水流过,或者可以在洗涤介质中制浆并且借助压滤器、倾析或离心经受再次分离。通常如此频繁地重复该过程,直至洗涤介质的电导率下降至某个值之下。这典型地为0.5ms/cm,尤其是0.2ms/cm。电导率根据din 38404第8部分来测定。
41.经分离和任选经洗涤的沉淀物的干燥在50至150℃范围内、优选70至130℃范围内、特别优选80至120℃范围内的温度进行。在此可以在喷雾干燥器中进行干燥。或者,也可以在固定式烘箱中进行干燥,在这种情况下,干燥时长通常在30分钟至6h范围内。
42.然后使经干燥的粉末经受煅烧。这在介于200与800℃之间,优选介于400与800℃之间发生,特别优选介于600与800℃之间,更优选介于700与800℃之间的温度下。煅烧的时长为介于30min与4h之间,优选介于1与3h之间,并且特别优选2h。
43.在一个实施方案中,随后使经干燥和经煅烧的沉淀物经受成型方法。在此,将根据步骤d)获得的沉淀物经受以下步骤:
44.e)将根据步骤d)的经煅烧的沉淀成型,以获得成型体。
45.常规的成型方法是压片、挤出和造粒。在一个优选的实施方案中,将经煅烧的沉淀物压片。
46.压片通常用压片机,例如kilian pressima型压片机进行。优选在添加润滑剂如石墨、油或硬脂酸盐/酯,优选石墨下进行压片。为此,将在步骤d)中获得的经煅烧的沉淀物与至少一种润滑剂混合,任选地压实和/或造粒,然后压片。润滑剂在混合物中的份额通常为介于0.5至5.0重量%之间,优选介于1.0与4.0重量%之间,基于待压片的物料的总重量计。
47.在一个实施方案中,将粘结剂添加至待成型的沉淀物。原则上适合作为粘结剂的是提高成型体的机械稳定性的所有化合物。合适的粘结剂是氧化铝,例如拟勃姆石、勃姆石或刚玉、二氧化硅、铝酸钙、硅酸钙或粘土矿物例如膨润土。
48.在一个实施方案中,将钙铝酸盐用作粘结剂。这是含有氧化物和/或氢氧化物形式的ca和al的化合物。例如包括通式xcao
·
yal2o3的烧结钙铝酸盐或通式ca
x
aly(oh)z的化学沉淀钙铝酸盐。但是取决于铝钙酸盐的处理,也可以存在在这两个经验公式之间的中间阶段,其同样适合作为粘结剂。除了这些元素之外,钙铝酸盐中还可以存在另外的元素。在一个优选的实施方案中,钙铝酸盐以小于5.0重量%,优选小于1.0重量%,并且特别优选小于0.1重量%的重量份额含有另外的元素,基于钙铝酸盐的重量计。
49.在本发明中使用的钙铝酸盐的ca/al原子比可以变化,并且优选为介于0.1与3.5之间,还更优选介于0.3与2.0之间。
50.适合作为钙铝酸盐的是合成制备的材料。但也可以使用天然存在的铝钙酸盐,例如加藤石。
51.钙铝酸盐可以在用作粘结剂材料之前经受热处理(煅烧)。这在介于300与800℃之间,优选介于450与750℃之间并且特别优选介于450与650℃之间的温度发生。
52.通常将粘结剂以这样的量添加至混合物中,使得成型体中的粘结剂的含量在2至30重量%范围内,优选在2至10重量%范围内,并且特别优选在2至5重量%范围内,基于烧失之后的成型体的总重量计。
53.当将铝钙酸盐用作粘结剂时,优选将该混合物另外与水混合,以增强钙铝酸盐的粘结效果。优选地,将经热处理的钙铝酸盐添加至混合物。
54.与钙铝酸盐结合的成型体的钙含量优选在0.14至17.02重量%范围内,优选在0.14至5.67重量%范围内,并且特别优选在0.14至2.84重量%范围内,基于烧失之后的成型体的总重量计。
55.当将钙铝酸盐用作粘结剂时,成型体中的钙铝酸盐的重量份额能够借助x射线衍射法测定。在此,使样品在bruker公司的d4 endeavor中在5至90
°
2θ范围内进行测量(步序列0.020
°
2θ,每步测量时间为1.5秒)。作为辐射,使用cukα1辐射(波长40kv,35ma)。样品台在测量期间以30转/min的速率绕其轴旋转。借助rietveld精修对获得的反射强度光谱进行定量分析,并确定样品中的钙铝酸盐的份额。为了测定各个晶相的份额,使用bruker公司的topas软件。
56.然后可以使在步骤e)中获得的成型体经受热处理。在此,使根据步骤e)获得的成型体经受以下步骤:
57.f)使成型体在介于200与800℃之间的温度经受30min至4h时长的热处理。
58.在一个实施方案中,在介于400与700℃之间的温度进行热处理。在另一实施方案中,该热处理的时长为介于1与3h之间并且特别优选介于1.5与2.5h之间。
59.可以使通过根据本发明的方法可获得的催化剂在用于催化反应之前才在另外的
步骤中还原。
60.在此优选通过在还原性气氛中加热催化剂而进行还原。还原性气氛尤其是氢气。例如在150℃至450℃范围内、优选在160℃至250℃范围内、特别优选在170℃至200℃范围内的温度进行还原。例如在1小时至20天的时间段内,优选在2小时至120小时的时间段内,特别优选在24至48小时的时间段内进行还原。在一个优选实施方案中,还原在190℃至210℃范围内的温度进行24至48小时的时间段。
61.在一个优选的实施方案中,使催化剂在还原之后以湿法或干法稳定化。在湿法稳定化的情况下,将催化剂用液体覆盖以尽可能减少与氧气的接触。合适的液体包括有机液体和水,优选有机液体。优选的有机液体是在20℃具有0.5hpa或更低的蒸气压的那些。这样的合适的有机液体的实例是异癸醇、nafol、脂肪醇、十六烷、2-乙基己醇、丙二醇及其混合物,特别是异癸醇。在干法稳定化的情况下,将氧气或含氧气体,优选空气和惰性气体如氩气或氮气的混合物计量添加至还原腔室中。混合物中的氧气的浓度优选从约0.04体积%提高至约21体积%。例如,可以计量添加空气和惰性气体的混合物,其中空气比惰性气体的比例最初为约0.2体积%的空气比99.8体积%的惰性气体。然后逐渐增加空气比惰性气体的比例(例如连续或逐步),直到最终例如计量添加100体积%的空气(对应于约21体积%的氧气浓度)。不受任何理论约束,假设通过计量添加空气或氧气,在催化剂表面处产生具有例如0.5至50nm、优选1至20nm、特别优选1至10nm厚度的薄氧化物层,其保护催化剂成型体免于另外的氧化。在干式稳定化的情况下,反应器温度优选为100℃以下,特别优选为20℃至70℃,并且最优选为30℃至50℃。可以在填充有催化剂的反应装置中非原位或原位进行还原。
62.片料形式的催化剂成型体的侧压强度在还原之后具有50至250n,优选60至200n,特别优选70至150n的值。
63.根据本发明的催化剂具有对含酸介质或含水介质,如具有作为杂质的酸和/或水的有机溶液或有机气态混合物的改进的稳定性。除了催化剂成型体的改进的侧压强度之外,这还表现对催化活性至关重要的材料中铜离子的减少的损失。此外,根据本发明的催化剂成型体还具有较低的金属离子总损失,这是固体结构针对单个金属离子浸出的提高的稳定性的标志。
64.为了测定根据本发明的催化剂成型体对酸影响的稳定性,使成型体在含酸和含水介质中经受处理,随后测定经如此处理的成型体的侧压强度以及在含酸和含水介质中金属离子的份额。
65.本发明的另一主题是根据本发明的催化剂用于催化氢化有机化合物中的羰基基团的方法,所述氢化在含酸和/或含水介质中进行。可能的反应包括将醛氢化成醇,尤其是将氧代醛氢化成氧代醇,将脂肪酸氢化,例如通过酯化,尤其是氢化成脂肪酸甲酯,和随后的氢解,或将酮氢化成相应的醇。
66.在此使用的反应介质的典型酸值在0.1至3.4mg
koh
/g
溶液
范围内,优选在0.2至1.0mg
koh
/g
溶液
范围内。酸值是酸性oh基团存在的量度,例如在羧酸中,在溶液中,并且能够例如通过用koh溶液滴定相应溶液至中和点来测定。在这种情况下消耗的koh量,基于溶液的形态,对应于酸值,以mg
koh
/g
溶液
表示。
67.这样的反应介质中的水含量通常在0.1至5.0重量%,优选0.2至5.0重量%,更优
选0.5至5.0重量%,特别优选0.5至3.0重量%范围内。
68.本发明范围内的待氢化的脂肪酸为饱和或不饱和脂肪酸,由于其链长而被分为短链(至多6-8个c原子)、中链(6-8直至12个c原子)和长链(13至21个c原子)。此外也可以使用具有多于22个c原子的脂肪酸。
实施例
69.在本发明的范围内,根据din 51081测定烧失量,这通过测定约1-2g待分析材料的样品的重量,然后在室气氛下将其加热至900℃并且在此温度下储存3h来进行。然后将样品在惰性气氛下冷却并且测量残余重量。热处理之前和之后的重量差异对应于烧失量。
70.侧压强度(scs)的测定根据astm 04179-01来进行而不预干燥片料。在此测量统计学上足够数量的片料(至少20个片料)并且计算各个测量值的算术平均值。该平均值对应于所测定的样品的侧压强度。
71.化学元素的测定按照din en iso 11885借助icp测量(电感耦合等离子体)来进行。
72.通过如下测定酸值:将约4g样品溶液与25ml丙醇混合,添加酚酞作指示剂。将溶液在室温用氢氧化四丁基铵溶液(0.1mol/l,于2-丙醇/甲醇中)滴定直至颜色骤变。按照下式计算以mg
koh
/g
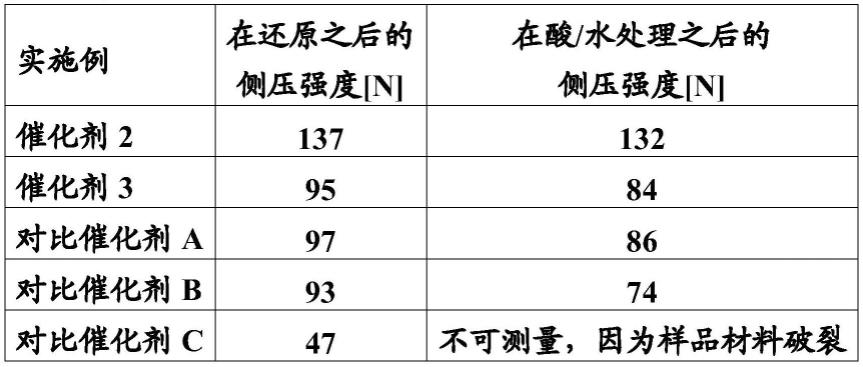
溶液
计的酸值sz:
[0073][0074]
其中,sz=酸值,消耗=以ml计的四丁基氢氧化铵溶液的消耗量,c=四丁基氢氧化铵溶液的浓度,m=koh的摩尔质量,和称量=以g计的所使用的样品溶液的量。
[0075]
催化剂成型体的孔体积根据din 66133按照汞孔隙测定法在1至2000巴的压力范围内测量。
[0076]
溶液的水含量根据astm e 203(2016)借助于karl fischer法测定。
[0077]
实施例1:根据本发明的催化剂1的制备
[0078]
通过将3530g cu(no3)2·
3h2o和1843g(zro)2(oh)2co3加至5000ml软化水中来制备水溶液1。随后通过添加1550ml硝酸实现盐的完全溶解。将酸性溶液用软化水补足至20 000ml的总体积。溶液的ph为-0.70。随后将溶液加热至80℃。
[0079]
此外,将1500g na2co3和2140g naalo2溶于22 000ml软化水,溶液2的ph值为12.23。
[0080]
为了沉淀,提供沉淀容器,其装有8000ml软化水。同时向其中引入含铜溶液1以及含碳酸盐溶液2。在此如此调节计量添加速率,使得沉淀溶液中存在约6.5的ph。
[0081]
在结束添加和完全沉淀之后,将沉淀物滤出和用软化水洗涤,直至洗涤水具有低于0.25ms的电导率,以除去粘附的杂质。随后将滤饼干燥。
[0082]
随后将经干燥的粉末在750℃煅烧2h。
[0083]
相对重量份额为cu=29.9重量%、zr=17.5重量%和al=20.6重量%,基于烧失之后的总质量计。
[0084]
实施例2:根据本发明的催化剂2的制备
[0085]
将1706g在实施例1中获得的经煅烧的粉末与51g secar 71粘结剂(31重量%的cao、69重量%的al2o3)、5g软化水和34g石墨合并,并且混合10分钟,从而产生均匀的混合物。将该混合物首先压实和造粒,然后在kilian公司的pressima型压片机中压制成具有4.5mm的宽度和3mm的高度的片料。随后使片料在600℃经受2h的煅烧。如此获得的片料的堆积密度为1175g/l。对于用途实施例3和4,根据相同工序制备片料,不同之处在于,片剂具有3.0mm的高度和同样3.0mm的宽度。片料中的相对重量份额为cu=29.0重量%、zr=17.0重量%、al=21.1重量%和0.6重量%的ca,基于烧失之后的总质量计。
[0086]
实施例3:根据本发明的催化剂3的制备
[0087]
将1706g在实施例1中获得的经煅烧的粉末与5g软化水和34g石墨合并,并且混合10分钟,从而产生均匀的混合物。将该混合物首先压实和造粒,然后在kilian公司的pressima型压片机中压制成具有4.5mm的宽度和3mm的高度的片料。随后使片料在600℃经受2h的煅烧。片料中的相对重量份额为cu=29.9重量%、zr=17.5重量%和al=20.6重量%,基于烧失之后的总质量计。
[0088]
对比实施例1(对比催化剂a)
[0089]
通过以下制备催化剂a:沉淀含铜和含铬的沉淀物,通过热处理转变成氧化形式和压制成具有4.5mm的宽度和3mm的高度的片料。相对重量份额为cu=37.5重量%和cr=23.0重量%,基于烧失之后的总质量计。对于用途实施例3和4,根据相同工序制备片料,不同之处在于,片剂具有3.0mm的高度和同样3.0mm的宽度。
[0090]
对比实施例2(对比催化剂b)
[0091]
为了制备催化剂b,通过将1250g cu(no3)2·
3h2o、220g mn(no3)2·
4h2o和1800g al(no3)3·
9h2o溶于9000g蒸馏h2o中制备水溶液1。通过将1720g na2co3溶于7500g蒸馏h2o中制备溶液2。将两个溶液单独在搅拌下加热至80℃。然后在连续搅拌下将两个溶液计量添加至沉淀容器中。将在此沉淀出的沉淀物滤出并且用蒸馏h2o洗涤,直至洗涤水具有低于0.25ms的电导率,以除去粘附的杂质。随后干燥滤饼。随后将经干燥的粉末在750℃热处理3h;相对重量份额为cu=44.8重量%、mn=7.0重量%和al=17.92重量%,基于烧失之后的总质量计。
[0092]
将1706g该粉末与51g secar 71粘结剂、5g软化水和34g石墨合并,并且混合10分钟,从而产生均匀的混合物。将该混合物首先压实和造粒,然后在kilian公司的pressima型压片机中压制成具有4.5mm的宽度和3mm的高度的片料。随后使片料在600℃经受2h的煅烧。片料中的相对重量份额为cu=43.5重量%、mn=6.8重量%、al=18.5重量%和ca=0.6重量%的,基于烧失之后的总质量计。
[0093]
对比实施例3(对比催化剂c)
[0094]
用于催化剂c的粉末根据用于催化剂b的粉末的制备方式来制备,其中如此选择mn(no3)2·
4h2o的份额,使得在如此获得的粉末中的锰的相对重量份额基于在烧失之后的质量计为0.1重量%。相对重量份额为cu=49.7重量%、mn=0.1重量%和al=20.0重量%,基于烧失之后的总质量计。将1706g如此获得的粉末与5g软化水和34g石墨合并,并且混合10分钟,从而产生均匀的混合物。将该混合物首先压实和造粒,然后在kilian公司的pressima型压片机中压制成具有4.5mm的宽度和3mm的高度的片料。片料中的相对重量份额为cu=49.7重量%、mn=0.1重量%和al=20.0重量%,基于烧失之后的总质量计。如此获得的片
料的堆积密度为1152g/l。
[0095]
使对比催化剂a、b和c以及根据本发明的催化剂2和3的在压片之后获得的材料的一部分经受还原。在这种情况下使样品在2体积%的h2和98体积%的n2的气体混合物中在200℃的温度进行热处理,以使存在的cuo还原成cu。随后将样品在氮气下冷却至室温并且储存在液态异癸醇下。随后测量该样品的侧压强度并且将其用于用途实施例1至3。
[0096]
用途实施例1(稳定性测试)
[0097]
对于根据本发明的催化剂2和3中的每一个以及对比催化剂a、b和c进行酸稳定性的测定,通过将总量为25g的经压片、还原和稳定化的样品与具有75g氧代醛溶液、1重量%水份额和0.2mg
koh
/g
溶液
酸值的液体混合物合并。将这样的混合物在氮气气氛下在120℃加热4天。将经压片的样品在结束测试时与液体混合物分离。然后立即测量其侧压强度。
[0098]
在进行测试之后,分析氧代醛溶液中是否存在cu、al、cr、mn和zr。
[0099]
表1:催化剂的侧压强度
[0100][0101]
借助表1明显可看出,根据本发明的催化剂2和3的侧压强度在还原之后已经高于由现有技术已知的催化剂。借助在测试结束之后的侧压强度的值更清楚地显示了对酸影响和水影响的提高的稳定性。在此,根据本发明的催化剂2另外具有最高的侧压强度值,而相比之下,不含铬的cualmn催化剂的片料,即对比催化剂c,在测试期间破裂并且不可能对侧压强度进行有意义的测量。
[0102]
表2:在稳定性测试之后的测试溶液中的金属浓度
[0103][0104]
来自表2的数据显示,根据本发明的催化剂在剧烈的测试条件下对于铜物质的损
失基本上是稳定的,而这对于对比催化剂而言明显更高。
[0105]
这些结果说明了通过将锆添加至含铜催化剂中所实现的有益效果,即对于酸和水的提高的稳定性,这表现在更高的机械稳定性和更低的催化剂本身的金属损失。
[0106]
用途实施例2:将氧代醛氢化成氧代醇
[0107]
将具有100ml体积的还原和湿法稳定化形式的根据本发明的催化剂2的床层装入反应器中,并且在氮气流下加热至在120至180℃范围内的温度,其中在该温度选择2天的反应时长。随后引导含有45重量%的醛、25重量%的相应醇和30重量%的副产物(链烷烃、烯烃等)并具有0.7重量%的水份额和0.2的酸值的液相通过反应器。以气相色谱法研究反应器下游产物流的成分。在相应温度时的整个运行时间内计算的产物流中的转化率和醇含量,如表3中所示。
[0108]
为了对比,使对比催化剂a和对比催化剂b的各个样品经受相同的条件,由此实现的结果同样示于表3中。
[0109]
表3:在不同温度时的醛氢化的转化率和醇含量
[0110][0111]
借助表3可以清楚地看出,根据本发明的催化剂在与市售含铬催化剂a在大致对应的相当的测试条件下实现了醛的转化。对于相应醇的形成也显示出类似的行为。因此,根据本发明的催化剂是迄今为止使用的含铬催化剂的环境友好型替代品。
[0112]
数据还表明,尽管对比催化剂b实现了相当的转化率,并且甚至明显改进的醇的形成,然而由于其低的物理稳定性使其不适合在剧烈的反应条件下长时间使用。
[0113]
同样将对比催化剂c用于相同的测试。然而,催化剂颗粒在测试期间分解,从而意味着不可能就醛转化率和醛选择性做出任何有意义的阐述。
[0114]
用途实施例3:将脂肪酸氢化,例如通过酯化和随后的氢解(fame)
[0115]
将具有5ml体积的根据本发明的催化剂2的床层以经还原的和湿法稳定化的形式装入反应器,并且随后计量添加200ml月桂酸甲酯,其水份额为0.062重量%和酸值为0.351mg
koh
/g
溶液
。然后将反应器气密密封并且在氮气流下加热至280℃的温度。随后通过阀门将压力为175巴的水流引入反应器,直到氮气被完全置换。然后用搅拌器将处于反应器中的氢气与月桂酸甲酯溶液流化,以使用催化剂进行氢化。
[0116]
通过出口阀定期取出溶液的样品,并且以气相色谱法研究其成分。由此给出的月桂酸甲酯的转化率值、1-十二烷醇的选择性和产率示于表4中。
[0117]
为了对比,使体积为5ml的对比催化剂a的床层经受相同的条件下,由此实现的结果同样示于表4中。
[0118]
表4:氢化月桂酸甲酯的反应数据
[0119]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。