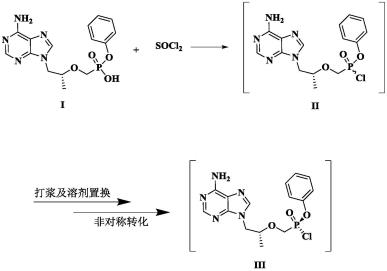
一种高非对映异构体膦酰氯的制备方法
(一)技术领域
1.本发明属于药物化学领域,具体涉及丙酚替诺福韦关键中间体单苯基替诺福韦膦酰氯进行非对称转化时提高转化率及反应速率的方法。
(二)
背景技术:
2.l-丙氨酸,n-[(s)-[[(1r)-2-(6-氨基-9h-嘌呤-9-基)-1-甲基乙氧基]甲基]苯氧基氧膦基]-,1-甲基乙基酯,(2e)-2-丁烯二酸(2:1),通用名为丙酚替诺福韦半富马酸盐(tenofovir alafenamide hemifumarate),其化学结构式如下
[0003][0004]
丙酚替诺福韦半富马酸盐(taf)是由吉利德科学有限公司研究开发的一种新型核苷类逆转录酶抑制剂(nrti),是比替诺福韦酯(tdf)更新一代的替诺福韦(tenofovir)前药。临床数据显示:在清除病毒能力效果相当的情况下,taf剂量更小,副作用低,具有更高的安全性与耐受性。其在生物利用度、毒性及临床给药剂量方面具有显著的优势,加上近年来药监部门对其的连续批准上市,其用于治疗hiv的taf复方制剂、三方制剂、单方制剂等陆续分别通过了fda、ema的批准,taf有望取代tdf,成为吉利德公司巩固其在感染性治疗领域领导者地位的利器。
[0005][0006]
丙酚替诺福韦化合物的合成过程如上所示:以pmpa为起始物料,先接上苯氧基得到中间体单苯基替诺福韦(式i),再连接l-丙氨酸异丙酯得到游离碱丙酚替诺福韦(gs-7340),最后与富马酸成盐得到taf。
[0007]
目前国内外已公开的由单苯基替诺福韦制备丙酚替诺福韦游离碱(gs-7340)的大致方法分为两种:1、先合成混合物的游离碱gs-7171(s
p
/r
p
≈50/50),在通过物理或者化学拆分得到高纯度的游离碱gs-7340;2、通过不对称合成得到高纯度s
p
构型的单苯基膦酰氯(式iii),再与l-丙氨酸异丙酯衍生反应使构型传承得到游离碱gs-7340。由于途径2方法具有更高的分子利用率,成为合成taf的首选路线。
[0008][0009]
cn103842366a公开了化合物i和氯化亚砜在甲苯介导下非对称合成得到非对映体比例s
p
:r
p
>90:10的中间体单苯基膦酰氯(iii),再与l-丙氨酸反应、精制得到高纯度游离碱gs-7340。在该方法中,非对称转化反应需要连续反应48~96小时,长时间的反应限制生产的效率和经济性,不利于工业化生产。
[0010][0011]
cn110981911a中还公开了另一种非对称合成高非对映异构体单苯基替诺福韦膦酰卤(s
p
/r
p
≥90%)的方法,即中间体单苯基替诺福韦在卤代试剂及有机溶剂a存在条件下先发生卤代反应,得到外消旋体的单苯基替诺福韦膦酰卤,再在有机溶剂b介导下发生非对称转化得到两种非对映异构体比例s/r≥90%的高非对映异构体单苯基膦酰卤,再与l-丙氨酸异丙酯反应,处理后得到高纯度的gs-7340。但是该方案非对称转化反应时间长,非对映异构体比例s/r最高只能达到92.0%。
[0012][0013]
作为非对称合成路径中关键中间体,高非对映异构体单苯基膦酰氯(式iii)合成时的转化率及反应速率,决定了后续衍生反应合成的游离碱丙酚替诺福韦的非对映异构体纯度及收率,极大地影响着原料药的质量和经济效益;作为本发明领域技术人员很容易理解的是,基于单苯基膦酰氯固有的化合物特性,无论是直接进行非对称合成制备高非对映异构体纯度的单苯基膦酰氯,还是先发生卤代反应制备单苯基膦酰氯消旋体,再进行非对称转化制备高非对映异构体单苯基磷酰氯,合成高非对映异构体膦酰氯的反应均是一个非均相反应过程,反应过程中固相单苯基膦酰卤的形态及介导溶剂b的组分,决定了非对称合成反应的的转化率及反应速度。基于单苯基替诺福韦膦酰氯化学性质十分活泼,在多数质子性溶剂中均不稳定,与水份接触容易水解等特性。在目前报道的相关合成工艺中,尚未有对该非均相反应中的固相形态进行改良以提高固相反应颗粒物的均一性及反应接触面的方法,也未确定非对称合成过程中介导溶剂组分的控制,使得合成高非对映异构体单苯基膦酰氯的转化率相对偏低,反应速率较慢,非对称转化反应产物中两种非对映异构体比例s
p
/r
p
仅能维持在90:10左右,非对称转化反应工艺的稳定性也存在一定的挑战和困难。工艺稳定性及分子利用率有待进一步提高。
(三)
技术实现要素:
[0014]
本发明的目的是提供一种单苯基替诺福韦膦酰氯制备方法,以显著提高非对称转化反应的反应速率。
[0015]
为实现上述发明目的,本发明采用如下技术方案:
[0016]
一种高非对映异构体膦酰氯的制备方法,包括如下步骤:
[0017]
1)式i所示的单苯基替诺福韦与氯化亚砜在溶剂a中发生卤代反应生成式ii所示的单苯基替诺福韦膦酰氯外消旋体,充分反应后所得反应混合物去除溶剂a,得到单苯基替诺福韦膦酰氯消旋体的固体或浆液;所述溶剂a选自乙腈、二氯甲烷、甲苯中的一种或多种的任意组合;
[0018]
2)步骤1)得到的单苯基替诺福韦膦酰氯消旋体的固体或浆液,加入无水溶剂c进行搅拌打浆,得到均一固体浆液;所述无水溶剂c选自无水二氯甲烷、乙腈、甲苯、二甲苯、乙基苯、环己烷、正己烷、正庚烷、乙醚、异丙醚、甲基叔丁基醚、环戊基甲醚中一种或多种溶剂任意组合;
[0019]
3)步骤2)得到的单苯基替诺福韦膦酰氯消旋体的浆液用无水溶剂b进行置换,得
到均一的单苯基替诺福韦膦酰氯消旋体的浆液;所述的无水溶剂b是单苯基替诺福韦膦酰氯消旋体经不对称转化得到高非对映异构体含量的单苯基替诺福韦膦酰氯的反应使用的介导溶剂,置换后浆液中溶剂a和溶剂c残留含量(气相色谱gc含量,该溶剂面积占溶剂总面积的百分比)分别不超过10%,进一步优选不超过5%,更进一步优选不超过2%;
[0020]
4)步骤3)所得单苯基替诺福韦膦酰氯消旋体的浆液,经无水溶剂b介导的不对称转化反应得到高非对映异构体含量的单苯基替诺福韦膦酰氯,所得高非对映异构体膦酰氯中两种非对映异构体比例s
p
/r
p
≥92%;
[0021][0022]
本发明步骤1)所述卤代反应可参照文献报道的反应条件进行,该反应不需要加入催化剂,通常当反应物剩余《1%时停止反应。作为优选,所述氯化亚砜的用量为化合物i摩尔当量的2~6倍。作为优选,所述溶剂a选自乙腈和/或二氯甲烷。更进一步优选所述的溶剂a为乙腈,所述卤代反应在50℃~回流温度下进行,反应时间为2h~6h。更进一步优选所述的溶剂a为二氯甲烷,所述卤代反应在回流温度下进行,回流时间为4~7小时。本发明特别优选所述卤代反应在乙腈中进行。本发明优选在卤代反应完成后,通过蒸馏浓缩去除溶剂a,得到单苯基替诺福韦膦酰氯消旋体的固体或浆液。
[0023]
本发明步骤2)中,所述无水溶剂c与化合物i的质量比为2-30:1,优选2-10:1,进一步优选5-10:1。打浆温度选自0℃~100℃间任意温度,优选打浆温度为0-60℃,进一步优选打浆温度为20℃~60℃;打浆时间选自0.5~24小时,优选打浆时间0.5~2小时,进一步优选打浆时间为1小时。特别优选所述无水溶剂c为无水二氯甲烷。
[0024]
本发明步骤3)中,置换方法具体为:蒸除步骤2)得到的单苯基替诺福韦膦酰氯消旋体的浆液中的溶剂,然后加入无水溶剂b搅拌均匀蒸馏至固体或浆状,加入无水溶剂b搅拌均匀蒸馏的操作可重复,直至溶剂a及c残留量达到所要求的水平。
[0025]
本发明步骤3)中无水溶剂b的选择和步骤4)中溶剂b介导的不对称转化的反应条件均可参考cn110981911a,这两者的控制对于获得高非对映异构体含量的单苯基替诺福韦
膦酰氯具有重要影响。
[0026]
作为优选,所述溶剂b为甲苯,所述不对称转化的反应温度为85~105℃。
[0027]
作为优选,所述溶剂b为二甲苯,所述不对称转化的反应温度为85~120℃。
[0028]
作为优选,所述溶剂b为乙基苯,所述不对称转化的反应温度为85~115℃。
[0029]
作为优选,所述溶剂b为氯苯,所述不对称转化的反应温度为90~120℃。
[0030]
最优选所述的不对称转化在无水甲苯中于85~105℃的温度条件下进行。在所述优选的溶剂b和反应温度条件下,反应2~14小时即能完成反应,其中,反应2小时后两种非对映异构体比例就能达到s
p
/r
p
≥92%;延长反应时间可以进一步提高单苯基膦酰氯的非对映异构体纯度,在非对称转化反应4小时至14小时内非对映异构体比例稳定维持在s
p
/r
p
≥93%,甚至达到>94%。
[0031]
本发明使用的无水溶剂可由工业级溶剂经除水处理后获得。
[0032]
与现有技术相比,本发明的有益效果在于:
[0033]
本发明方法操作简单,可控性强,实施后可大大缩短非对称转化反应的时间,并提高单苯基替诺福韦膦酰氯的非对映异构体纯度,工艺稳定性好,适合丙酚替诺福韦半富马酸盐的工业化生产。
[0034]
具体而言,单苯基替诺福韦膦酰氯是一种十分活泼的氯代物,接触空气水分和或质子性溶剂会迅速水解或反应,并且氯代反应生产过程通常因蒸馏出的单苯基膦酰氯固体颗粒性状各异而导致后续转化反应的转化速率和非对映异构体比例波动较大。本发明采用不亲水的非质子溶剂打浆,避免了氯代物因引入水分而发生分解或反应,在保证物料稳定前提下,通过打浆步骤改善了单苯基替诺福韦膦酰氯在非均相的非对称转化反应过程固相物料的均一性及反应接触面,从而显著提高了非对称转化反应的反应速率,使单苯基膦酰氯非对称转化反应在反应2小时后两种非对映异构体比例就能达到s
p
/r
p
≥92%;并且延长反应时间可以进一步提高单苯基膦酰氯的非对映异构体纯度,在非对称转化反应4小时至14小时内非对映异构体比例稳定维持在s
p
/r
p
≥93%,甚至可达到高于94%,从而降低后续去除对映异构体杂质的工艺难度及复杂程度,提高分子利用率。适合丙酚替诺福韦半富马酸盐的工业化生产。
(四)附图说明
[0035]
图1a和1b分别是实施例1氯代反应得到的化合物ii的外消旋体hplc检测得到的对映体比例和含量。
[0036]
图2a和2b分别是实施例1不对称转化2h时hplc检测得到的对映体比例和含量。
[0037]
图3a和3b分别是实施例1不对称转化4h时hplc检测得到的对映体比例和含量。
[0038]
图4a和4b分别是实施例1不对称转化14h时hplc检测得到的对映体比例和含量。
[0039]
需要说明的是,图谱中保留时间11分钟处的峰为残留的转化溶剂甲苯峰,不计入含量。本发明在对不对称转化产物进行hplc检测前,需要对产物进行吹干处理以去除甲苯,由于吹干程度不同,故hplc检测得到的甲苯残留量存在差异。
(五)具体实施方式
[0040]
以下通过具体实施例详细说明本发明实施过程和产生的有益效应,旨在帮助阅读
者更好地理解本发明的实质和特点,不作为对本方案可实施范围的限定。
[0041]
实施例1至实施例6为一种高非对映异构体膦酰氯的制备方法的具体实施例;对比例1和对比例2为卤代反应蒸馏结束后打浆与不进行打浆之间的对照实验;对比例3至对比例5为卤代反应蒸馏结束后选用不同溶剂打浆的对照实施例。
[0042]
实施例1
[0043]
1)室温下,氮气保护下向250ml反应器中投入化合物i(10g,0.028mol),乙腈(77ml),氯化亚砜(8.2g,0.069mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hpl c分析ii的s
p
:r
p
=49:51);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状或者固体;
[0044]
2)向体系中加入无水二氯甲烷(50g),升温至40℃左右回流搅拌打浆1小时,得到化合物ii颗粒分布均一的浆液;再次缓慢蒸馏浓缩去除二氯甲烷,得到化合物ii的浆液;
[0045]
3)向体系中加入无水甲苯(80g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、二氯甲烷,得到化合物ii的固体;
[0046]
4)向体系中加入无水甲苯(150g,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.04%,二氯甲烷含量0.33%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量97.0%,非对映异构体比例s
p
/r
p
为92.3%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量97.1%,非对映异构体比例s
p
/r
p
为94.0%;保温反应时间达14h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量96.4%,非对映异构体比例s
p
/r
p
为94.3%。
[0047]
实施例2
[0048]
1)室温下,氮气保护下向500ml反应器中投入化合物i(15g,0.041mol),乙腈(115ml),氯化亚砜(12.28g,0.103mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49:51);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状或者固体;
[0049]
2)向体系中加入无水二氯甲烷(150g,10w),升温至40℃左右回流搅拌打浆1小时,得到化合物ii颗粒分布均一的浆液,再次缓慢蒸馏浓缩去除二氯甲烷,得到化合物ii的浆液或固体;
[0050]
3)向体系中加入无水甲苯(120g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、二氯甲烷,得到化合物ii的浆液或固体;
[0051]
4)向体系中加入无水甲苯(225g,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.35%,二氯甲烷含量1.7%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量97.5%,非对映异构体比例s
p
/r
p
为92.0%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量97.1%,非对映异构体比例s
p
/r
p
为93.7%;保温反应时间达到13h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量97.2%,非对映异构体比例s
p
/r
p
为94.2%。
[0052]
实施例3
[0053]
1)室温下,氮气保护下向500ml反应器中投入化合物i(20g,0.055mol),乙腈(154ml),氯化亚砜(16.4g,0.137mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=48.5:51.5);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应
体系呈浆状或者固体;
[0054]
2)向体系中加入无水甲苯(160g,8w),升温至60℃左右保温搅拌打浆1.5小时,得到化合物ii颗粒分布均一的浆液,再次缓慢蒸馏浓缩去除甲苯,得到化合物ii的浆液;
[0055]
3)向体系中加入无水甲苯(160g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈,得到化合物ii的浆液或固体;
[0056]
4)向体系中加入无水甲苯(300g,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.1%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量96.8%,非对映异构体比例s
p
/r
p
为92.9%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量96.6%,非对映异构体比例s
p
/r
p
为93.9%;保温反应时间达到12h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量97.1%,非对映异构体比例s
p
/r
p
为94.0%。
[0057]
实施例4
[0058]
1)室温下,氮气保护下向500ml反应器中投入化合物i(15g,0.041mol),乙腈(115ml),氯化亚砜(12.3g,0.103mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=48.5:51.5);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0059]
2)向体系中加入无水正庚烷(150g,10w),升温至60℃左右搅拌打浆1.5小时,得到化合物ii颗粒分布均一的浆液,再次缓慢蒸馏浓缩去除正庚烷,得到化合物ii的浆液;
[0060]
3)向体系中继续加入无水甲苯(120g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、正庚烷,得到化合物ii的固体;
[0061]
4)向体系中加入无水甲苯(225g,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.05%,正庚烷含量0.7%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量95.7%,非对映异构体比例s
p
/r
p
为92.2%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量96.8%,非对映异构体比例s
p
/r
p
为93.1%;保温反应时间达到14h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量95.5%,非对映异构体比例s
p
/r
p
为93.4%。
[0062]
实施例5
[0063]
1)室温下,氮气保护下向500ml反应器中投入化合物i(15g,0.041mol),乙腈(115ml),氯化亚砜(12.3g,0.103mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=48.3:51.7);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0064]
2)向体系中加入无水甲基叔丁基醚(150g,10w),升温至55℃左右搅拌打浆1.5小时,得到化合物ii颗粒分布均一的浆液,再次缓慢蒸馏浓缩去除甲基叔丁基醚,得到化合物ii的浆液;
[0065]
3)向体系中继续加入无水甲苯(120g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、甲基叔丁基醚,得到化合物ii的固体;
[0066]
4)向体系中加入无水甲苯(225g,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.15%,甲基叔丁基醚含量0.5%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量94.2%,非对映异构体比例s
p
/r
p
为92.3%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量93.6%,非对映异构体比例s
p
/r
p
为93.1%;保温反应时间
达到12h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量93.6%,非对映异构体比例s
p
/r
p
为93.2%。
[0067]
实施例6
[0068]
1)室温下,氮气保护下向反应釜中投入化合物i(15kg,41.287mol),乙腈(115l),氯化亚砜(12.3kg,103.36mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49:51);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0069]
2)向体系中加入无水二氯甲烷(150kg,10w),升温至40℃左右回流搅拌打浆1小时,得到化合物ii颗粒分布均一的浆液,再次缓慢蒸馏浓缩去除二氯甲烷,得到化合物ii的浆液;
[0070]
3)向体系中继续加入无水甲苯(120kg,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、二氯甲烷,得到化合物ii的浆液;
[0071]
4)向体系中加入无水甲苯(225kg,15w)搅拌成均一浆状(gc分析体系中乙腈含量0.16%,二氯甲烷含量1.1%),将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量98.3%,非对映异构体比例s
p
/r
p
为92.1%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量98.5%,非对映异构体比例s
p
/r
p
为93.3%;保温反应时间达到13h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量98.5%,非对映异构体比例s
p
/r
p
为93.6%。
[0072]
对比实施例
[0073]
对比例1为按照专利cn110981911a实施例1所述方案,用3.0eq氯化亚砜进行氯代反应,反应结束后直接蒸除溶剂,不使用溶剂进行打浆及置换;非对称转化反应2小时,非对映异构体比例s
p
/r
p
为76.2%;反应13小时,非对映异构体比例s
p
/r
p
为92.0%。
[0074]
对比例2为专利cn110981911a实施例1所述方案的对照实验,用3.0eq氯化亚砜进行氯代反应,反应结束后用二氯甲烷进行打浆,甲苯进行置换。非对称转化反应2小时,非对映异构体比例s
p
/r
p
为92.6%,反应13小时,非对映异构体比例s
p
/r
p
为94.2%。
[0075]
对比例3至5为实施例1至6的对照实验,卤代反应结束后蒸馏去除反应溶剂乙腈,并选用本发明研究所认为的合适的打浆溶剂c以外的溶剂乙酸乙酯、丙酮、二甲基亚砜进行打浆,并用甲苯置换;非对称转化反应12小时,单苯基替诺福韦膦酰氯含量在89.3%~92.3%,非对映异构体比例s
p
/r
p
为82.8%~89.0%。
[0076]
对比例1
[0077]
室温下,氮气保护下向2000ml反应器中投入化合物i(100g,0.275mol),乙腈(1200ml),投入氯化亚砜(100g,0.826mol),升温至回流保温反应4小时,得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=48:52),减压蒸馏浓缩去除反应溶剂乙腈,得到单苯基替诺福韦膦酰氯,向体系中加入无水甲苯(1500ml),将体系升温至90℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量96.0%,非对映异构体比例s
p
/r
p
为76.2%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量96.2%,非对映异构体比例s
p
/r
p
为83.0%;保温反应时间达到13小时,结束反应,hplc检测单苯基替诺福韦膦酰氯含量96.0%,非对映异构体比例s
p
/r
p
为92.0%,此时非对映异构体比例达到峰值,再延长反应时间也不再增加。
[0078]
对比例2
[0079]
室温下,氮气保护下向2000ml反应器中投入化合物i(100g,0.275mol),乙腈(1200ml),投入氯化亚砜(100g,0.826mol),升温至回流保温反应4小时,得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49:51);减压蒸馏浓缩去除反应溶剂乙腈至体系呈浆状,投入无水二氯甲烷(1500g),40℃下保温搅拌1小时,得到化合物ii颗粒分布均一的浆液;减压蒸馏去除二氯甲烷,再次向体系中加入无水甲苯(1200g),60℃搅拌均匀后继续蒸馏去除乙腈和二氯甲烷,得到化合物ii的浆液;向体系中加入无水甲苯(1500ml)搅拌均匀呈均一浆状(gc分析检测体系中乙腈含量0.03%,二氯甲烷含量0.65%),缓慢升温至90℃保温搅拌反应2小时,hplc检测单苯基替诺福韦膦酰氯含量97.5%,非对映异构体比例s
p
/r
p
为92.6%;反应4小时,hplc检测单苯基替诺福韦膦酰氯含量97.3%,非对映异构体比例s
p
/r
p
为93.6%;反应13小时,hplc检测单苯基替诺福韦膦酰氯含量97.2%,非对映异构体比例s
p
/r
p
为94.2%。
[0080]
对比例3
[0081]
1)室温下,氮气保护下向500ml反应器中投入化合物i(15g,0.041mol),乙腈(115ml),氯化亚砜(12.3g,0.103mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49.5:50.5);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0082]
2)向体系中加入无水乙酸乙酯(150g,10w),升温至55℃左右搅拌打浆1小时,得到化合物ii浆液,再次缓慢蒸馏浓缩去除乙酸乙酯,得到化合物ii的浆液;
[0083]
3)向体系中继续加入无水甲苯(120g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、乙酸乙酯,得到化合物ii的固体;
[0084]
4)向体系中加入无水甲苯(225g,15w)搅拌成均一浆状,将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量92.6%,非对映异构体比例s
p
/r
p
为77.1%;保温反应4小时,hplc检测单苯基替诺福韦膦酰氯含量91.8%,非对映异构体比例s
p
/r
p
为79.6%;保温反应时间达到12h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量89.3%,非对映异构体比例s
p
/r
p
为82.8%。
[0085]
对比例4
[0086]
1)室温下,氮气保护下向500ml反应器中投入化合物i(20g,0.055mol),乙腈(154ml),氯化亚砜(16.4g,0.138mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49.4:50.6);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0087]
2)向体系中加入无水丙酮(150g,7.5w),升温至55℃左右搅拌打浆1小时,得到化合物ii浆液,再次缓慢蒸馏浓缩去除丙酮,得到化合物ii的浆液;
[0088]
3)向体系中继续加入无水甲苯(160g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、丙酮,得到化合物ii的固体;
[0089]
4)向体系中加入无水甲苯(300g,15w)搅拌成均一浆状,将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量90.8%,非对映异构体比例s
p
/r
p
为60.0%;保温反应时间达4小时,hplc检测单苯基替诺福韦膦酰氯含量90.9%,非对映异构体比例s
p
/r
p
为74.6%;保温反应时间达到14h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量90.0%,非对映异构体比例s
p
/r
p
为86.1%。
[0090]
对比例5
[0091]
1)室温下,氮气保护下向500ml反应器中投入化合物i(20g,0.055mol),乙腈(154ml),氯化亚砜(16.4g,0.138mol),升温至70℃保温反应4h,溶清得到化合物ii的外消旋体(hplc分析ii的s
p
:r
p
=49.1:50.9);减压蒸馏,缓慢浓缩去除反应溶剂乙腈,直至反应体系呈浆状;
[0092]
2)向体系中加入无水二甲基亚砜(150g,7.5w),升温至60℃左右搅拌打浆1.5小时,得到化合物ii浆液,再次缓慢蒸馏浓缩去除二甲基亚砜,得到化合物ii的浆液;
[0093]
3)向体系中继续加入无水甲苯(160g,8w),60℃搅拌均匀并再次缓慢蒸馏浓缩去除甲苯及残留溶剂乙腈、二甲基亚砜,得到化合物ii的固体;
[0094]
4)向体系中加入无水甲苯(300g,15w)搅拌成均一浆状,将体系升温至95℃保温搅拌反应2h,hplc检测单苯基替诺福韦膦酰氯含量93.1%,非对映异构体比例s
p
/r
p
为80.7%;保温反应时间达到4小时,hplc检测单苯基替诺福韦膦酰氯含量92.9%,非对映异构体比例s
p
/r
p
为86.6%;保温反应时间达14h,结束反应,hplc检测单苯基替诺福韦膦酰氯含量92.3%,非对映异构体比例s
p
/r
p
为89.0%。
[0095]
表1
[0096]
[0097]
[0098]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。