1.本发明涉及使用生物相容性共聚物作为用于活性剂递送的载体来进行活性剂(例如,药物)的递送,所述生物相容性共聚物包含侧链连接的氨基酸,所述活性剂直接与所述侧链连接的氨基酸结合或通过连接基团分子与所述侧链连接的氨基酸结合。
背景技术:
2.癌症是人类健康的主要威胁之一,并且鉴于事实上其可能性与年龄有关,因此病例数将随着人口老龄化而增加。berger,na等人,(2006)cancer in the elderly,transactions of the american clinical and climatological association 117:147-156;yancik,r(2005)cancer j.11:437-41。近年来,由于肿瘤特异性试剂(例如单克隆抗体)的使用,肿瘤疗法已经取得了巨大的进步。这些抗体阻断增殖信号,例如表皮生长因子途径(egfr)(西妥昔单抗(cetuximab),merck kgaa;/帕尼单抗(panitumumab),amgen/曲妥珠单抗(trastuzumab),roche);或者通过靶向血管内皮生长因子(vegf)途径(贝伐单抗(bevacizumab),roche)以减慢肿瘤生长来阻止新血管的形成。由于它们的靶抗原通常在肿瘤组织中过度表达,因此健康细胞受到的较小的损害,因此与传统的细胞毒性剂相比,抗体疗法具有较少的脱靶效应。zhou,q.(2017)biomedicines 5(4);reichert,jm(2017)mabs 9:167-181。抗体的独特特异性还用于旨在将细胞毒性药物靶向到肿瘤细胞的组合方法。这些所谓的抗体药物缀合物(adc)已被证明优于使用抗体或细胞毒性剂的单一疗法。尽管自二十世纪六十年代就广为人知,但adc概念直到最近才在制药行业中引起了兴趣,并且超过60种adc正在进行临床试验。mullard,a(2013)nat rev drug discov 12:329;beck,a等人,(2017)nat rev drug discov 16:315-337。
3.第一代adc使用抗体中的游离氨基拉附接细胞毒性药物和药物连接基团构建体。每个抗体具有多达80个游离氨基,它们的不加选择的官能化会导致由于细胞毒性药物意外附接至抗体的结合界面而具有不同的药物与抗体比率(dar)和亲和力的高度异质adc种类。通过调节反应中所使用的药物和抗体的化学计量,可以在一定程度上限制dar的异质性。关于位点特异性,异质性仅受进行首次临床试验时的化学可及性的限制。又花了另外20年的时间fda才批准了首批adc。由于以下原因,adc的发展已显著增加:30种adc已进入反应组。后一种异质性也是首批adc的主要问题和监管问题。yao,h等人,(2016)int j mol sci 17(2):194。此外,首批adc基于已知会引起重要免疫应答的小鼠免疫球蛋白。由于这些缺陷,第一代adc未能显示出相对于传统疗法的大量改善,因此作为首批被fda批准的adc的吉妥珠单抗(gemtuzumab)奥佐米星在2010年由pfizer公司自愿退出市场。beck,a等人,(2017)nat rev drug discov 16(5):315-337;beck,a等人,(2010)discov med 10(53):329-39。
4.第二代adc通过靶向人源化抗体的游离硫醇基来减轻后一种困难。这些游离硫醇
基是在偶联反应之前通过抗体铰链区中的4个链间二硫键的温和还原(例如,使用1,4-二硫苏糖醇(dtt))生成的。使用这种策略,可以将潜在的附接位点减少至8个,从而导致adc的更高同质性。考虑到事实上链间二硫键在抗体完整性中起着至关重要的作用,较高的同质性通常是为对抗体稳定性的负面影响而付出的代价。尽管设计了更特别的连接基团来保持二硫键完整性(如例如在shaunak,s等人,(2006)nat chem biol 2(6):312-3和balan,s等人,(2007)bioconug chem 18(1):61-76中详细说明的),但是所生成的adc遭受了通常为约3-4的低dar。如果进一步增加载药量,则抗体的稳定性会受到不利影响,从而导致从血流中快速清除。另外,抗体对其肿瘤细胞特异性靶标的亲和力受到了负面影响。beck等人,(2017)nat rev drug discov 16(5):315-337;yao等人,(2016)int j mol sci 17(2):194.;beck等人,(2010)discov med 10(53):329-39。由于只有少数细胞毒性实体与这些抗体偶联,常规的细胞毒性剂如多柔比星被证明在杀伤肿瘤细胞方面不够有效。tolcher,aw(1999)j clin oncol 17(2):478-478。因此,必须使用新颖类别的细胞毒性剂,所述细胞毒性剂的细胞毒性要高几个数量级。这些物质的示例是微管抑制剂,例如美登素(mertansine)(dm1)或单甲基澳瑞他汀(monomethylauristatin)e(mmae)。beck等人(2017)。对于此类有效药物,至关重要的是药物只能在它们的靶位点处从adc中释放。否则,可能会导致严重的副作用。药物与抗体之间的连接基团由此起着主要作用。最近投入市场的adc,如曲妥珠单抗emtansine(roche)和本妥昔单抗(brentuximab)vedotin(tekada pharmaceutical)以及mersana概念(mersana therapeutics inc.(cambridge,ma))使用基于马来酰亚胺的连接基团,已知所述连接基团与带有半胱氨酸的蛋白质,特别是血清白蛋白反应。alley,sc等人,(2008)bioconjug chem 19(3):759-765。shen,bq等人,(2012)nat biotechnol 30(2):184-9。
5.所谓的第三代adc利用药物与抗体的位点特异性偶联。一个著名的示例是来自seattle genetics的抗急性髓性白血病(aml)的vadatuximab tailirine。adc在两条重链的239位处都含有经遗传工程改造的半胱氨酸,所述经遗传工程改造的半胱氨酸用于偶联能够交联dna的吡咯并苯并二氮杂卓(pbd)二聚体,从而阻断细胞分裂并导致细胞死亡。adc已在i期研究中成功测试,并且目前正处于iii期临床试验中。beck等人,(2017);kennedy,da等人,(2015)cancer res 75(15增刊),摘要ddt02-04。药物与抗体的位点特异性偶联的其他示例使用智能标签,例如“醛标签”(redwood biosciences,catalent)或“分选酶标签”(smac-technology
tm
,nbe therapeutics;stefan,n等人,(2017)mol cancer ther 16(5):879-892)。后两种方法在抗体中引入了经遗传工程改造的肽标签,以作为酶促偶联反应的特定动机。第三代adc代表了具有增加的稳定性,但仍每种抗体仅递送少量毒性实体的更均质的产品。
6.为了避免这种限制,mersana therapeutics最近开发了一种使用聚合物载体的新颖方法。该概念基于具有几种细胞毒性药物分子的可降解载体聚合物(称为“fleximer”)的官能化。随后,通过常规连接基团化学物将载有药物的聚合物偶联至单克隆抗体。使用此,dar可以增加到每个抗体分子12-15个药物分子,所述药物分子分布在3-5个附接的聚合物载体上。“non-clinical pharmacokinetics of xmt-1522,a her2 targeting auristatin-based antibody drug conjugate”;poster presentation at the american association for cancer research(aacr)annual meeting in washington d.c,2017。尽
管此方法具有许多优点,但是所得adc含有可变链长和载药量的fleximer聚合物。与所使用的硫醇-马来酰亚胺连接基团化学组合时,adc的分子量在一定程度上变化。此外,fleximer聚合物包含可生物降解的酯键,从而带来了长期存储和/或血清稳定性的问题。koitka,m等人,(2010)j pharm biomed anal 51(3):664-78;li,b等人,(2005)biochem pharmacol 70(11:1673-84。
7.除抗体外,还详细说明了用于阻断或激活异常途径以治疗代谢性疾病和癌症的其他靶标特异性试剂(包括适体)。适体是具有通过沃森-克里克碱基配对(watson-crick base-pairing)形成的限定3维构象的小单链多核苷酸。由于它们的明确限定的结构,可以使它们以高亲和力结合特定靶标,包括分离的小分子,例如细菌毒素或细胞表面标记。mercier,mc等人,(2017)cancers(basel)9(6):e69;ruscito,a等人,(2016)front chem 4:14。适体远小于抗体,更易于生产并且缺乏免疫原性。ray,p等人,(2013)archivum immunologiae et therapiae experimentalis 61(4):255-271;pei,x等人,(2014)mol clin oncol 2(3):341-348;zhou,g等人,(2016)oncotarget7(12):13446-63。它们通常是在涉及迭代结合、洗涤和扩增步骤的富集过程中,从具有多达10
15
个随机多核苷酸的库生成的。在每个循环之后,选择具有最高靶标亲和力的适体用于下一个循环。这导致在10-12个循环后选择出了在纳摩尔或甚至亚纳摩尔范围内具有结合亲和力的分子。该过程也称为通过指数富集(selex)进行配体的系统进化。zhou,g等人(2016)。类似于抗体,第一治疗性适体方法旨在通过与关键蛋白、受体或代谢物的相互作用来阻断疾病相关的途径。一个著名的示例是(哌加他尼钠(pegaptanib);eyetech pharmaceuticals,pfizer),其为2004年上市的首个被fda批准的适体治疗剂。是27个核苷酸长的rna适体,并且用于年龄相关性黄斑变性(amd),amd是一种导致失明的严重眼病。amd的特征在于由于生长因子水平升高而异常形成血管。的靶标是vegf
165
(同种型),一种负责血管生成的生长因子。因为该适体由于快速的肾脏清除和降解而仅具有短半衰期,因此将其与40kda peg聚合物结合以增加其总体大小。另外,一些核苷酸被用2'-氟-嘧啶和2'-o-甲基-嘌呤取代,以避免被核酸酶降解。biagi,c等人,(2014)eur j clin pharmacol 70(12):1505-12;pozarowska,d等人,(2016)cent eur j immunol 41(3):311-316。与抗vegf抗体(例如,贝伐单抗,roche)不同,由于在全身性应用中的不良表现而从未用于或获得许可用于癌症治疗,所述不良表现可能是由于旁路途径(例如,pdgf-b)的效应的补偿。alvarez,rh等人,(2006)mayo clin proc 81(9):1241-57。随着近年来做出的改进,进行了数种尝试以将适体不仅用于靶向和阻断,而且还用作细胞毒性剂的载体。bagalkot及其同事开发出了适体-多柔比星复合物。然而,该复合物遭受了不良的负载效率和快速的系统清除。bagalkot,v等人,(2006)angew chem int ed 45(48):8149-8152。在2010年,基于载有多西紫杉醇/顺铂的plga-peg纳米粒子,开发了一种不同的方法。通过用靶向肿瘤细胞膜蛋白的适体a10进行官能化,将这些粒子导向至前列腺癌细胞。这种相当复杂的药物递送系统至少在体外实验中显示出了有前途的结果。kolishetti,n等人,(2010)proc natl acad sci usa 107(42):17939-17944。进一步测试了适体对几种基于核苷酸的治疗剂例如sirna(通常设计用于抑制特异性基因表达的短干扰rna)的递送。chu,tc等人,(2006)nucleic acids res 34(10):e73。尽管开发了许多不同的利用适体的靶向能力进行
肿瘤治疗的方法,但是直到目前,适体仍具有较差的负载能力、血清不稳定和快速的肾清除率,所有这些特性限制了它们的临床应用。这些适体-药物缀合物或复合物均未进入临床iii期或市场。zhou等人,(2016)。
8.为了在保持抗体/适体对相应靶标的亲和力的同时克服上述缺点并增大药物与抗体/适体的比率(dar),开发了一种新策略,该新策略利用生物相容性、亲水性、不可降解的共聚物作为活性剂载体。可以使聚合物携带多种(在极限内,任何期望的数量)活性剂分子。可以将所述共聚物偶联至靶向肿瘤的部分,例如单克隆抗体或适体。这种偶联可以发生在用活性剂或其它有效负载装载共聚物之前或之后。由于共聚物的高亲水性,所述共聚物能够携带甚至高度疏水的细胞毒性药物,同时保持相应抗体/适体的药代动力学特性。
9.本公开中提出的方法的优点在于,仅需要一个偶联位点即可将多个活性剂分子结合至抗体或适体分子。通过使用位点特异性偶联方法,例如与抗体重链c末端处的肽标签的酶促偶联反应,含活性剂的共聚物将定位在距抗体的结合界面适当距离处。使用这种方法,保留了对靶组织的最大亲和力,并且获得了相对均质的产品。所选择的连接策略在共聚物与抗体/适体之间形成了稳定的肽键,这确保了adc在血流中的高度稳定性。此外,所选择的共聚物设计有助于两种或更多种不同活性剂与同一共聚物分子的偶联,从而使得能够进行组合疗法。一旦活性剂(例如,在癌症情境下的细胞毒性药物)在靶向的细胞(例如,肿瘤细胞)内释放并且靶向部分(例如,抗体或适体)被降解,则据信相对较小的共聚物通过肾脏清除而被从体内清除。
技术实现要素:
10.本公开涉及一种含有多个第一有效负载分子的分子的共聚物分子,并且涉及一种用于制备该共聚物的方法。承载有效负载的共聚物是通过以下方式制备的:
11.(a)使反应混合物聚合,所述反应混合物包含
12.(1)含有叠氮基部分的式i的共-主要单体,
[0013][0014]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-;l是在生理条件下可分裂或不可分裂的连接基团/间隔基团。
[0015]
(2)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸部分或叠氮基部分,和
[0016]
(3)用于生成自由基的引发剂体系,聚合产生共聚物;以及
[0017]
(b)通常通过点击反应,将通常用点击反应性基团官能化的第一有效负载分子偶联至步骤(a)的共聚物中包含的叠氮基部分。
[0018]
术语“氨基酸部分”是指氨基酸通过其反应性侧链与丙烯酸部分偶联,并含有官能
化或非官能化的α-氨基和α-羧基官能团。术语“叠氮基部分”是指叠氮基。
[0019]
优选地,x部分通过其-nh-、-o-或-s-基团附接至c(o)-部分。
[0020]
在本发明的范围内,“用于生成自由基的引发剂体系”并不意味着被特别限制,并且可以使用技术人员已知的任何此类体系。本文描述了所述示例。该术语还意味着涵盖使用紫外线辐射来生成自由基。
[0021]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0022]
上述反应混合物可进一步包含:式ii的共-主要单体,
[0023][0024]
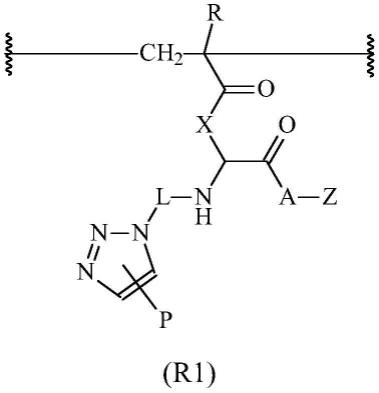
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;y为h或-co-c
nh2n 1
(其中n=1-8);z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-,
[0025]
和/或式iii的共-主要单体,
[0026][0027]
其中:r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;z为h(如果a为o)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-。
[0028]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0029]
式i的示例性不可分裂的连接基团/间隔基团l可以是-co-c
nh2n
(其中n=1-10)或-co-pegn(其中n=1-14)。优选地,pegn在本文中被理解为根据式-(och2ch2)
n-或-(ch2ch2o)
n-的部分。式i的可分裂连接基团/间隔基团l包括组织蛋白酶b敏感性连接基团,例如二肽连接基团、-co-缬氨酸-瓜氨酸-pabc或其变体、缬氨酸-赖氨酸、缬氨酸-丙氨酸、缬氨酸-精氨酸或三肽连接基团(例如谷氨酸-缬氨酸-瓜氨酸)、ph敏感性连接基团(例如腙或顺式基于乌头基的连接基团)、用于溶酶体运输和分裂的连接基团(例如焦磷酸二酯)。pabc是对苯胺-β-氨基甲酸酯。变体在本文中优选被定义为其中瓜氨酸被另一个氨基酸残基取代的部分。
[0030]
包含式i以及式ii和/或iii的单体的共聚物可以装载两种不同的有效负载。将通
常用点击反应性基团官能化的第一有效负载与式i的单体的叠氮基部分反应。通常被衍生为含有合适反应性基团的第二有效负载接合至式ii和iii的单体的氨基酸部分的α-氨基或α-羧基。这要求在式ii和/或iii的单体的至少一部分中为α-氨基或α-羧基或者未官能化的(游离的),即在式ii的单体中y或z中的一者或两者是h,和/或在式iii的单体中z是h。步骤(a)的共聚物可以首先装载第一有效负载,随后装载第二有效负载。或者,步骤(a)的共聚物可以首先装载第二有效负载,随后装载第一有效负载。因此,承载有效负载的共聚物的制备涉及聚合步骤(a)、偶联步骤(b)和进一步的步骤,在所述进一步的步骤中含有反应性基团的第二有效负载分子偶联至式ii和/或iii的共-主要单体。后一步骤可以发生在步骤(b)之前或之后。
[0031]
取决于活性剂的结构,活性剂分子可以直接或通过连接基团结构间接地与共聚物中式i的共-主要单体的叠氮基或式ii和/或iii的共-主要单体的α-氨基或α-羧基反应。后者连接基团应在存储期间和在血流中保持稳定,以避免意外释放细胞毒性药物。所述连接基团可能能够被特定的细胞内酶分裂,或者可能是“不可降解的”类型并且仅在具有溶酶体和过氧化物酶体的恶劣环境中被破坏。
[0032]
在优选的实施方案中,(步骤(a)的)共聚物是通过反应混合物的聚合来制备的,所述反应混合物进一步包含用于控制共聚的raft试剂。raft试剂可含有2-30个单位的单分散间隔基。此外,raft试剂可以以反应性基团为特征,所述反应性基团可例如用于附接至具有细胞类型特异性或组织类型特异性靶向部分的共聚物。后一反应性基团可以是硫醇、醛、炔烃、叠氮化物、胺、羧基、酯、双吖丙啶、苯基叠氮化物、硫酯、重氮、施陶丁格反应性膦酸酯(或膦基硫酯)、肼、肟、用于执行氮杂迈克尔连接的丙烯酸酯、或能够用于酶促偶联反应中的基序。所述基序可以是包含2-8个氨基酸的寡聚甘氨酸(所述肽基序使得能够进行分选酶介导的偶联反应)、转谷氨酰胺酶反应性底物、醛标签、自催化内含肽序列、或点击反应性基团。或者,raft试剂可能能够在聚合后转化,以提供用于将细胞类型特异性或组织类型特异性靶向部分附接至共聚物的反应性基团。通常,一旦聚合和/或官能化已完成,raft试剂就会失活,由此通过热处理、与合适的胺反应(氨基分解)、或在磷含氧酸存在下与引发剂分子发生新反应或在不存在磷含氧酸的情况下与过量引发剂发生新反应,来执行raft基团的消除。
[0033]
共聚物的制备(步骤(a))也可涉及两个连续的聚合反应。第一反应混合物可包含不含氨基酸基团或叠氮基部分的可聚合主要单体、用于控制共聚的raft试剂、和用于生成自由基的引发剂体系,所述聚合产生了raft预聚物。所述第二聚合反应在第二反应混合物中进行,所述第二反应混合物包含所述第一聚合反应的所述raft预聚物、式i的共-主要单体和用于生成自由基的引发剂体系。第二反应混合物可进一步包含式ii和iii中的一者或两者的共-主要单体和/或不含氨基酸部分或叠氮基部分的可聚合主要单体。
[0034]
本公开的共聚物通常进一步用细胞类型特异性或组织类型特异性靶向部分官能化。靶向部分可以在用第一和/或第二有效负载分子官能化之前或之后,即在步骤(b)或涉及将第二有效负载分子偶联到式ii和/或iii的共-主要单体的步骤之前或之后,附接至共聚物。
[0035]
潜在的靶向部分为但不限于单克隆抗体、抗体片段、纳米抗体(单结构域抗体)、darpin(经设计的锚蛋白重复蛋白)、肽激素、与肿瘤细胞表面上表达的蛋白质结合的蛋白
质、基于dna或rna的适体,或能够与已知在肿瘤细胞中过表达的细胞表面受体(例如叶酸或生物素)结合的小分子。靶向部分的共价附接是以位点特异性方式进行的,通常涉及在共聚物的头基中的反应性基团(通常经由raft试剂或通过转化所述raft试剂引入)。合适的偶联策略包括所谓的“点击化学反应”,所述“点击化学反应”在共聚物的头基处使用反应性基团(例如在[3 2]环加成的情况下使用应变炔烃,或在[4 2]环加成的情况下使用应变烯烃),所述头基随后用于结合含有点击反应的“配对部分”(例如对于[3 2]环加成是叠氮基,或对于[4 2]环加成是四嗪)的细胞类型特异性或组织类型特异性靶向部分。点击反应的上述反应性部分意谓为可互换的。通过使用正交相容的点击反应,例如[3 2]环加成和[4 2]环加成反应的组合,用于用有效负载分子修饰共聚物并将靶向部分附接至所述共聚物的“点击反应”可以以顺序方式或“并行”地进行。
[0036]
通常,用点击反应的配对部分来修饰靶向部分应该以定点方式执行,例如,通过使用酶促偶联技术(如转谷氨酰胺酶介导的或分选酶介导的偶联)或者通过在合成期间或合成后将非典型(非天然)氨基酸整合到靶向部分中。
[0037]
在不同的实施方案中,酶促偶联反应也可用于用含有多个有效负载分子的共聚物直接修饰细胞型或组织型特异性靶向部分。这是通过用合适的标签(例如用于分选酶介导的偶联的寡聚甘氨酸、醛标签或转谷氨酰胺酶标签)修饰聚合物的头基来实现的。在转谷氨酰胺酶介导的反应的情况下,由合适的链转移剂引入的共聚物的头基可包含含有反应性赖氨酸(或谷氨酰胺)残基的肽基序,或非肽基序,例如含有末端氨基的连接基团结构。后一头基修饰可以尤其与微生物转谷氨酰胺酶组合使用,已知所述微生物转谷氨酰胺酶以高周转率接受非肽基序。
[0038]
有效负载分子可以是活性剂或螯合剂。在其中有效负载是螯合剂的本发明的承载有效负载的共聚物的情况下,将所述共聚物与能够被螯合剂捕获的活性剂一起孵育或暴露于所述活性剂。优选地,其中所述共聚物暴露于活性剂的情况是指如本文所公开的将所述共聚物与活性剂接触。在短寿命放射性同位素的情况下,使用螯合剂作为有效负载分子可能是特别有利的,所述短寿命放射性同位素可以在诊断或治疗性使用之前立即结合到承载有效负载的共聚物。
[0039]
为了清楚起见,上面和权利要求中使用的术语,例如“式i的共-主要单体”、“主要单体”、“式ii的共-主要单体”、“raft试剂”和“引发剂体系”并不意味着指一种类型的分子数。单数也意味着包括复数。例如,术语“式i的共-主要单体”是指存在各种量的满足式i的要求的一种或多种化学上不同的化合物。
[0040]
在上述携带多个有效负载分子的共聚物的任何共聚物中,所述共聚物的平均分子量为5,000道尔顿至80,000道尔顿。更优选地,所述共聚物的平均分子量为5,000道尔顿至40,000道尔顿。最优选地,所述共聚物的平均分子量为5,000道尔顿至20,000道尔顿。这些分子量不包括偶联的靶向部分的重量。本发明中共聚物的平均分子量优选被理解为数均分子量,所述数均分子量是基于尺寸排阻色谱法(size exclusion chromatography,sec)/凝胶渗透色谱法(gel permeation chromatography,gpc)的测量,通过与已知的分子量标准品(在本文中为不同的支链淀粉聚合物)进行比较来计算的。详情参见实施例13中呈现的方法。作为替代或补充,在上述承载多个有效负载分子的共聚物中的任何共聚物中,至少80%(w)的共聚物分子的平均分子量为5,000道尔顿至80,000道尔顿。更优选地,所述共聚物分
子的至少80%(w)的平均分子量为5,000道尔顿至40,000道尔顿。最优选地,所述共聚物分子的至少80%(w)的平均分子量为5,000道尔顿至20,000道尔顿。因此,最后一段中呈现的百分比部分是共聚物的多分散指数(polydispersion index,pdi)的结果。分散性(也称为多分散性指数(pdi)或异质性指数)是给定聚合物样本中分子量分布的度量。聚合物的(pdi)由以下公式定义:pdi=mw/mn,其中mw是重均分子量并且mn是数均分子量,如通过凝胶渗透色谱法使用已知聚合物作为参考标准品所测量。分散性表示一批聚合物中单独分子质量的分布。本发明中共聚物的pdi通常在1.03至1.4的范围内,优选地在1.05至1.35的范围内,更优选地在1.1至1.30的范围内,最优选地在1.15至1.25的范围内。除非另有说明,否则pdi的特征在于实施例13中呈现的方法。
[0041]
优选地,在含有式i的共-主要单体作为唯一共-主要单体的共聚物中,共-主要单体的平均数为2-12。更优选地,所述共聚物平均含有2-8个共-主要单体,最优选2-6个共-主要单体。对于还含有未官能化的式ii和/或式iii的共-主要单体的共聚物,所有共-主要单体的优选平均数为10-50。更优选的是为10-40的共-主要单体平均数,最优选的是为10-30的共-主要单体平均数。如果式ii和/或式iii的共-主要单体被用有效负载分子官能化,则所有共-主要单体的优选平均数为4-20。更优选的是为4-15的共-主要单体平均数,最优选的是为4-10的共-主要单体平均数。如本文所理解的,术语“官能化的”涉及将第二有效负载分子附接至式ii和/或式iii的单体。如果单体未被官能化,则应理解为它们不含第二有效负载分子。换句话说,第二有效负载分子没有附接至所述单体。
[0042]
如果本公开的共聚物中所含有的有效负载分子是活性剂,则这种活性剂可以是微管抑制剂、嵌合剂、烷基化剂、抗代谢物、激素或激素受体调节剂、酪氨酸激酶抑制剂、能够干扰基因或其相应的信使rna的基于多核苷酸的药物、基于蛋白质的细菌毒素、适用于前药疗法的酶(adept概念)、或放射性同位素。活性剂也可以是示踪剂分子,所述示踪剂分子包括小分子荧光团、基于蛋白质/肽的荧光团、近红外(nir)荧光探针、生物发光探针、放射性对比剂或放射性同位素。
[0043]
本发明优选进一步涵盖一种共聚物,所述共聚物含有第一有效负载分子的多个拷贝,所述共聚物,可通过以下步骤获得:
[0044]
(a)使反应混合物聚合,所述反应混合物包含
[0045]
(1)式ii的单体,
[0046][0047]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x是-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;y是h;z是h(如果a是-o-)或-c
nh2n 1
,
其中n=1-8;并且a是-o-或-nh-,
[0048]
(2)具有至少一个乙烯基且不含氨基酸部分或叠氮基部分的单体,以及
[0049]
(3)用于生成自由基的引发剂体系,所述聚合产生共聚物;
[0050]
(b)用含有连接基团/间隔基团l和叠氮基部分的胺反应剂处理步骤(a)的所述共聚物,以及
[0051]
(c)将所述第一有效负载分子偶联至在步骤(b)的所述共聚物中包含的所述叠氮基部分。
[0052]
或者,y也可以被定义为h或-c
nh2n 1
b(其中n=1-8),其中b是h或oh,优选地y是h。如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0053]
上述反应混合物可以进一步优选地补充有式iii的单体,
[0054][0055]
其中:r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;z为h(如果a为o)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-。
[0056]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0057]
进一步优选地,本发明涉及一种包含式(r1a)的重复单元的共聚物
[0058][0059]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x是-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;z是h(如果a是-o-)或-c
nh2n 1
(其中n=1-8);并且a是-o-或-nh-,l是连接基团/间隔基团。l如本文所定义。如本文所定义的聚合物可根据本发明的方法获得。
[0060]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优
选地,如果a是-o-,则z优选是h。
[0061]
进一步优选地,本发明涉及一种包含式(r1)的重复单元的共聚物
[0062][0063]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x是-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;z是h(如果a是-o-)或-c
nh2n 1
(其中n=1-8);并且a是-o-或-nh-,l是连接基团/间隔基团,并且p包含第一有效负载分子。l和有效负载分子如本公开中所定义。
[0064]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0065]
优选地,包含式(r1a)的重复单元的共聚物或包含式(r1)的重复单元的共聚物进一步包含:式(r2)的重复单元:
[0066][0067]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x是-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;y是h或-co-c
nh2n 1
(其中n=1-8),或者y包含第二有效负载分子;z是h(如果a是-o-)或-c
nh2n 1
1(其中n=1-8),或者z包含第二有效负载分子;并且a是-o-或-nh-,
[0068]
和/或式(r3)的重复单元:
[0069][0070]
其中:r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;z为h(如果a是o)或-c
nh2n 1
(其中n=1-8);或者z包含第二有效负载分子;并且a为-o-或-nh-。
[0071]
如本文优选理解的,如果a是-o-,则z是h或-c
nh2n 1
(其中n=1-8),而如果a是-nh-,则z是-c
nh2n 1
(其中n=1-8)。或者,z也可以优选被定义为z是h或-c
nh2n 1
(其中n=1-8)。优选地,如果a是-o-,则z优选是h。
[0072]
优选地,z可以是h和/或y可以是h。或者,z和/或y可包含第二有效负载分子。有效负载分子如本文所定义。或者,在某些实施方案中,z是h或-c
nh2n 1
(其中n=1-8),或者z包含第二有效负载分子。
[0073]
进一步优选地,本发明的共聚物包含可通过n,n-二甲基丙烯酰胺、n-异丁基丙烯酰胺、n-叔丁基丙烯酰胺、n-羟乙基丙烯酰胺、n-(2-羟丙基)-丙烯酰胺、n-(3-羟丙基)-丙烯酰胺、n-(3-羟丙基)-甲基丙烯酰胺、n-(2-羟丙基)-甲基丙烯酰胺、n-(3-氨基丙基)-丙烯酰胺盐酸盐、或n-(3-氨基丙基)-甲基丙烯酰胺盐酸盐的聚合获得的重复单元,或者通过甲基丙烯酸、2-羟乙基丙烯酸酯、2-羟丙基丙烯酸酯、3-羟丙基丙烯酸酯、2-羟基-1-甲基乙基-丙烯酸酯、2-氨基乙基丙烯酸酯盐酸盐、3-羟丙基甲基丙烯酸酯、2-羟基-1-甲基乙基甲基丙烯酸酯、2-羟乙基甲基丙烯酸酯、2-羟丙基甲基丙烯酸酯或2-氨基乙基甲基丙烯酸酯盐酸盐的聚合获得的重复单元。
[0074]
进一步优选地,在本发明的共聚物中,其中不存在式(r2)和(r3)的重复单元,每分子共聚物中根据式(r1)的重复单元的平均数为2至12,优选地2至8,更优选地2至6。
[0075]
进一步优选地,在本发明的共聚物中,其中如本文所定义,所述式(r2)或(r3)的重复单元未被官能化,每分子共聚物中根据式(r1)、(r2)或(r3)的重复单元的平均数为10至50,优选地10至40,更优选地10至30。
[0076]
进一步优选地,在本发明的共聚物中,其中所述式(r2)或(r3)的重复单元被用第二有效负载分子官能化,每分子共聚物中根据式(r1)、(r2)或(r3)的重复单元的平均数为4至20,优选地4至15,更优选地4至10。
[0077]
本公开还涉及药物组合物,所述药物组合物包含有效量的本公开的共聚物,所述共聚物含有共价结合的活性剂或被共价结合的螯合剂捕获的活性剂的多个分子;以及载体。取决于活性剂的性质,这些组合物可用于治疗各种癌症或其他疾病/病症。
[0078]
本公开还涵盖治疗不同类型的癌症或其他疾病和病症的方法,所述方法包括施用药物组合物,所述药物组合物包含有效量的本公开的共聚物,所述共聚物含有共价结合的
活性剂或被共价结合的螯合剂捕获的活性剂的多个分子;和载体(所述共聚物在本文中也称为“活性部分”)。在本公开的范围内还有包含有效量的本公开的共聚物的药物组合物用于治疗受试者的癌症或另一种疾病或病症的用途,所述共聚物包含共价结合的活性剂或被共价结合的螯合剂捕获的活性剂的多个分子;和载体,所述用途包括向受试者施用有效量的共聚物。
[0079]
本发明进一步优选地涉及本发明的共聚物在疗法中的用途,其中所述共聚物包含第一有效负载分子和第二有效负载分子,并且其中所述第一有效负载分子和第二有效负载分子是要作为联合疗法一起施用的两种活性剂。
[0080]
本发明进一步优选地涉及本发明的共聚物,所述共聚物用于诊断应用,优选地用于癌症监测。优选地,癌症的监测是与癌症疗法同时执行的。优选地,如本文所公开的,所述共聚物包含可用于诊断的放射性核素。
具体实施方式
[0081]
除非另有定义,否则所有术语应具有它们在相关领域中的普通含义。定义了以下术语,并且以下术语将具有以下含义:
[0082]
如本文所用,“药学上可接受的载体或赋形剂”旨在包括与药物施用相容的任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗和吸收延迟剂等,例如无菌无热原水。合适的载体描述于本领域中的标准参考文献remington's pharmaceutical sciences(mack publishing co.,easton,pa,第19版,1995)中,该文献以引用方式并入本文。可以用作药学上可接受的载体的材料的非限制性示例是糖,例如乳糖、葡萄糖和蔗糖;环糊精,例如α-环糊精、β-环糊精和γ-环糊精;淀粉,例如玉米淀粉和马铃薯淀粉;纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素和乙酸纤维素;粉末状黄蓍胶;麦芽;明胶;滑石;赋形剂,例如可可脂和栓剂用蜡;油,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;二醇类,例如丙二醇;酯类,例如油酸乙酯和月桂酸乙酯;琼脂;缓冲剂,例如氢氧化镁和氢氧化铝;藻酸;无热原水;等渗盐水;林格氏溶液;乙醇和磷酸盐缓冲溶液,以及其他无毒的相容润滑剂,例如十二烷基硫酸钠和硬脂酸镁,并且根据配方师的判断组合物中也可以存在着色剂、释放剂、包衣剂、甜味剂、调味剂和芳香剂、防腐剂和抗氧化剂。还包含乳化剂/表面活性剂(例如cremophor el和solutol hs15)、卵磷脂和磷脂(例如磷脂酰胆碱)。也可以使用脂质体。用于药学活性物质的此类介质和药剂的用途是本领域中众所周知的。除非任何常规介质或试剂与活性化合物不相容,否则考虑将所述介质或试剂用于组合物中。补充的活性化合物也可以掺入组合物中。
[0083]
如本文所用,术语“受试者”是指哺乳动物受试者。优选地,所述受试者是人类受试者。
[0084]
术语“活性部分”涉及本公开的共聚物,所述共聚物含有有效负载分子的多个分子(所述共聚物可以进一步用细胞类型特异性或组织类型特异性靶向部分官能化),由此,如果有效负载分子是螯合剂,则该术语是指还含有已经被螯合剂捕获的活性剂的共聚物。
[0085]
在本公开的上下文中,术语“细胞类型特异性或组织类型特异性靶向部分”是指以亲合力结合特定类型的细胞或特定组织的细胞上的表面标记的分子,所述亲合力使所述分子可用于将货物活性剂递送至细胞中。它可以是单克隆抗体、抗体链的单结构域可变片段、
单链抗体、重复蛋白darpin(设计的锚蛋白重复蛋白)、基于dna或rna的适体、基于肽的适体、能够结合细胞表面标记的肽或蛋白质、激素、或能够结合细胞表面标记的小分子。
[0086]“示踪分子”被定义为能够在诊断或科学应用中产生读出信号的分子。它可以是小分子荧光团、基于蛋白质/肽的荧光团、近红外(nir)荧光探针、生物发光探针、放射性对比剂、或放射性同位素。
[0087]
本公开的活性部分的“有效量”是指活性部分当在治疗过程中施用一次或多次时,以适用于任何医疗治疗的合理益处/风险比赋予对被治疗的受试者的治疗效应的量。治疗效应可以是客观的(即,可通过某种测试或标记进行测量)或主观的(即,受试者给出指示或感觉到效应)。本公开的活性部分的有效量是包含活性剂的活性部分的量,优选为在约0.01mg/kg受试者体重至约50mg/kg受试者体重,并且更优选地约0.1mg/kg受试者体重至约30mg/kg受试者体重范围内的量。有效剂量还将取决于施用途径以及与其他试剂共用的可能性而变化。然而,应当理解的是,本公开的活性部分和药物组合物的总每日用量将由主治医师在合理的医学判断范围内决定。任何特定患者的具体有效剂量水平将取决于各种因素,包括正在治疗的疾患和疾患的严重程度;所采用的特定活性剂的活性;所采用的具体组成;患者的年龄、体重、总体健康状况、性别和饮食;所采用的特定活性部分的施用时间、施用途径和排泄速率;治疗的持续时间;与所采用的特定活性部分组合或同时使用的药物;以及医学领域中众所周知的类似因素。应注意,当在预防或防止的上下文中使用时,本公开的活性部分的“有效量”是指活性部分当在治疗过程中施用一次或多次时的赋予被治疗的受试者期望的预防效应的量。
[0088]
术语“有效负载分子”或“有效负载”是指直接或通过连接基团共价连接至共聚物的活性剂,或者是指共价连接至共聚物并且能够捕获活性剂的螯合剂。共价偶联到共聚物的螯合剂可用于固定化放射性同位素。螯合剂包括但不限于(1,4,7,10)-四氮杂环十二烷-1,4,7,10-四乙酸[dota]、2,2',2
”‑
(10-(2,6-二氧四氢-2h-吡喃-3-基)-1,4,7,10-四氮杂环十二烷-1,4,7-三基)三乙酸[dota-ga]、1,4,7-三氮杂环壬烷-n,n',n
”‑
三乙酸[nota]、1,4,8,11-四氮杂环十四烷-1,4,8,11-四乙酸)[teta]、二亚乙基-三胺-五乙酸酐[dtpa]、以及sarcophagine螯合剂如sar(参见nicholas等人,2019,angewandte chemie 131:15133-15136)。
[0089]
术语“活性剂”是指治疗活性物质,所述治疗活性物质与本公开的共聚物结合。在癌症疗法的情况下,活性剂通常是细胞毒性物质/分子。细胞毒性物质/分子的示例包括微管抑制剂,例如单甲基奥瑞斯他汀e(mmae)或emtansine(dm1);插入药物,例如多柔比星;烷基化剂,例如环磷酰胺(cp);抗代谢物,例如5-氟尿嘧啶(5-fu);激素或激素受体调节剂,例如枸椽酸他莫昔芬;酪氨酸激酶抑制剂,例如阿法替尼(afatinib)或博舒替尼(bosutinib);基于肽的毒素,例如α-鹅膏蕈碱;免疫检查点抑制剂,例如或适用于抗体指导的酶前药疗法(adept)的酶;能够干扰一种或多种基因或其相应信使rna(sirna、微rna或反义rna)的基于多核苷酸的药物;以及放射性同位素,例如但不限于氟-18、铜-64、镓-68、锆-89、铟-111、碘-123(诊断应用)或铜-67、锶-89、钇-90、碘-131、钐-153、镥-177、镭-223和锕-225(治疗性应用)。其他示例性活性剂包括钪-43、钪-44、铽-152和铽-155(诊断应用)或钪-47、铽-149和铽-161(治疗应用)。所列的放射性核素中的一些放射性核素适用于诊断和治疗应用。例如,铽-161可以用作治疗剂,但是由于其
γ-发射,在使用γ相机时也具有一定可见性,或者铽-149可以用于靶向α疗法并且在pet扫描中具有可见性。此外,本文提出的放射性核素也可以组合成“治疗诊断串联”以用于治疗方法中生物分布的可视化。
[0090]
术语“活性剂”还涵盖放射性同位素/放射性核素,所述放射性同位素/放射性核素通常借助于与共聚物的共-主要单体共价连接的螯合剂偶联至所述单体。
[0091]
本发明范围内的术语“放射性同位素/放射性核素”优选同义地使用,并且优选地代表具有过量核能,从而使其不稳定的原子。这种过量的能量可以以下三种方式中的一种使用:从原子核发出作为γ射线;转移到其电子中的一个电子上以将其作为转变电子释放;或者用来从原子核创建和发射新粒子(α粒子或β粒子)。放射性同位素/放射性核素在本文中优选被定义为半衰期小于10
19
年的同位素。
[0092]
术语“活性剂”还涵盖能够例如通过抑制抗凋亡因子例如bcl-2或靶向细胞外排泵(例如mdr-1转运体)来克服肿瘤细胞抗性的物质,或包括皮质类固醇、糖皮质激素和非甾体抗炎药(例如,前列腺素)在内的可用于减少与炎症相关的疗法副作用的抗炎物质。
[0093]
术语“胺反应剂”优选涉及可与氨基反应,从而通过共价键附接至其的部分。
[0094]
术语“拷贝”优选涉及分子的多个实例,即多于一个实例,其中该分子的多于一个实例具有相同的化学结构。
[0095]“单体”是指可聚合的低分子量化合物。对于式i至iii的共-主要单体,或对于主要单体,低分子量通常是指小于800道尔顿的分子量。当在共聚物的上下文中提及时,术语“单体”是指共聚物的最小结构单元。
[0096]
术语“重复单元”优选涉及在(均聚或共聚的)聚合物中重复多次的二价亚结构。通常,在可通过含有双键的单体的(共)聚合获得的聚合物中,重复单元的附接点对应于由所述双键连接的原子。换句话说,重复单元是通过将所述单体包含在聚合物中而衍生自所述单体的。
[0097]
术语“raft试剂”和“raft方法”优选地没有特别限制,并且可以指任何类型的可逆加成-片段化链转移。术语“raft试剂”和“raft方法”涉及在合适的链转移剂(cta)存在下单体的常规自由基聚合。常用的raft试剂包括硫代羰基巯基化合物,例如双硫酯、二硫代氨基甲酸酯、三硫代碳酸酯和黄原酸酯,这些试剂经由可逆链转移过程介导聚合。chiefari,j.等人,(1998)macromolecules 31(16):5559-62。
[0098]
术语“预聚物”涉及短聚合物,所述短聚合物以raft试剂为首,并且包含10-25个单元的亲水性主要单体,例如二甲基丙烯酰胺。此类预聚物代表水溶性大分子raft试剂,所述水溶性大分子raft试剂在第二聚合反应中用于在水性环境中合成主要单体与共-主要单体的共聚物。
[0099]
术语“底物、基序或标签”或“反应性底物,基序或标签”可互换使用,以涉及能够参与酶促催化反应的化学结构。这些化学结构可以被酶的活性中心识别,并且可以在发生酶促催化反应之前在中间形成共价或静电的酶-底物复合物。在本公开的上下文中,这些反应往往用于介导本公开的共聚物与肿瘤细胞特异性或组织特异性靶向部分的共价附接。典型的底物、基序和标签是共聚物的头基的柔性间隔基团中的氨基酸或肽、反应性官能团(如氨基、硫醇或羧基)或不饱和碳键的限定序列。
[0100]
术语“聚合物类似反应”优选地被定义为大分子链中承载的官能团的有意改变,其
总体目标是维持原始大分子的聚合度。
[0101]
术语“抗体-药物缀合物”,缩写为“adc”,表示靶向细胞类型特异性或组织类型特异性抗原(包括肿瘤抗原)的抗体与一种或多种药物分子的组合,其中所述药物分子共价附接至所述抗体。在本公开的上下文中,adc是指靶向细胞类型特异性或组织类型特异性抗原的抗体与本公开的展示多个活性剂分子的共聚物的缀合物。如所讨论的,本公开的共聚物承载有多个活性剂分子或不同活性剂分子的组合,所述活性剂分子通过连接基团或直接共价结合至共-主要单体中的叠氮基、α-氨基和α-羧基。它还可以承载大量螯合剂,所述螯合剂通过连接基团或直接共价结合至共-主要单体中的叠氮基、α-氨基和α-羧基,并且所述螯合剂捕获活性剂分子。
[0102]
术语“抗体放射性核素缀合物”(antibody radionuclide conjugate,arc)优选被定义为adc的变体,其中“药物分子或活性分子”代表共价结合到抗体-聚合物缀合物(例如在放射性碘的情况下)或者通过金属螯合剂复合物例如与放射性镥、锕或铽结合的放射性核素/放射性同位素。如此形成的arc能够向肿瘤组织递送大量辐射,从而由于dna、必需酶等的破坏而杀死肿瘤细胞。
[0103]
术语“适体”被定义如下:适体是与特定靶分子结合的寡核苷酸或肽分子。通常通过以下方式来创建适体:在迭代富集过程中从大型随机序列库中选择适体,以鉴定具有最高靶标亲和力的适体序列。该过程也称为“通过指数富集(selex)进行配体的系统进化”。更具体地,适体可以分类为dna、rna、异源核酸(xna)(在糖骨架方面不同的天然核酸合成替代物)或肽适体。适体由寡核苷酸链(通常为短链)或氨基酸序列组成。寡核苷酸序列可由此由一种类型的核苷酸(例如dna)或不同核苷酸类型的组合形成,所述不同核苷酸类型为例如dna、rna和/或特别设计的所谓“锁核苷酸(locked-nucleotide)”,所述锁核苷酸的核糖部分被用连接2'氧和4'碳的额外桥修饰。本公开中的适体还指由一个(或多个)短肽结构域组成的肽适体。
[0104]
术语“适体-药物缀合物”是指适体与一个活性剂分子或不同活性剂分子的组合。在本公开的上下文中,在将共聚物偶联至适体之前或之后,将活性剂分子附接至共聚物。
[0105]
术语“增强的渗透性和保留(epr)效应”用于描述肿瘤组织中的异常分子和流体传输动力学,尤其是关于大分子药物。某些大小的分子(通常是脂质体、纳米粒子和大分子药物)倾向于在肿瘤组织中以比正常组织中更高的水平积累。对于这种现象给出的一般解释是,为了使肿瘤细胞快速生长,它们必须刺激血管生成。新形成的肿瘤血管通常在形式和架构上是异常的,并且对于较高分子量的分子来说是可渗透的。此外,肿瘤组织通常缺乏有效的淋巴引流,使得一旦分子已经进入肿瘤组织,就不能有效地将所述分子从该组织中清除。
[0106]
在共-主要单体的上下文中,术语“侧链连接的氨基酸”是指氨基酸通过其侧链(例如,通过酯键或酰胺键)共价连接至含有丙烯酰基的部分。式i至iii的单体含有侧链连接的氨基酸。
[0107]
术语“主要单体”和“共-主要单体”主要用于促进本发明的描述。主要单体是指不包含氨基酸部分(即官能化或非官能化的氨基酸)或叠氮基的单体,并且共-主要单体是指含有氨基酸部分的单体。优选地,术语“主要单体”或“可聚合主要单体”可与术语“具有至少一个乙烯基且不含氨基酸部分或叠氮基部分的单体”或“不含氨基酸部分或叠氮基部分的单体”互换使用。优选地,术语“主要”和“主”在本文中可互换使用。
[0108]
可存在于本发明的共-主要单体中的侧链连接的氨基酸包括赖氨酸(k)、酪氨酸(y)、丝氨酸(s)、苏氨酸(t)、半胱氨酸(c)、4-羟基脯氨酸(ho-p)、鸟氨酸(orn)和4-氨基-苯丙氨酸(hox)。氨基酸可以是l形式或d形式,或外消旋混合物。在共聚物中,可以存在单一类型的侧链连接的氨基酸或多种类型的侧链连接的氨基酸。例如,共聚物可包含丙烯酰基-l-赖氨酸(ak)和丙烯酰基-l-苏氨酸(at)两者。为了清楚起见,由式i至iii描述的所有单体包含侧链连接的氨基酸。式i的单体在氨基酸的α-氨基处被用叠氮基官能化。式ii和iii的单体可以是未官能化的,或者可以在它们含有的氨基酸部分的α-氨基和/或α-羧基处被官能化。本公开的含氨基酸的共聚物包含一种或多种可聚合的主要单体,所述单体的特征为具有至少一个乙烯基但不含氨基酸残基或叠氮基残基;一种或多种根据式i中的任一者的共-主要单体;以及任选的一种或多种式ii和/或式iii的共-主要单体。
[0109]
优选地,在仅含有式i的共-主要单体的共聚物中,共-主要单体的平均数为2-12。更优选地,所述共聚物平均含有2-8个共-主要单体,最优选2-6个共-主要单体。对于还含有未官能化的式ii和/或式iii的共-主要单体的共聚物,所有共-主要单体的优选平均数为10-50。更优选的是为10-40的共-主要单体平均数,最优选的是为10-30的共-主要单体平均数。如果式ii和/或式iii的共-主要单体被官能化,则所有共-主要单体的优选平均数为4-20。更优选的是为4-15的共-主要单体平均数,最优选的是为4-10的共-主要单体平均数。
[0110]
先前描述了含有侧链连接的氨基酸的单体的合成。zbaida,d等人,(1987)reactive polymers,ion exchangers,sorbents 6(2-3):241-253。可以通过以下方式来制备此类单体:使赖氨酸、酪氨酸、丝氨酸、苏氨酸、半胱氨酸、鸟氨酸、4-氨基-苯丙氨酸或4-羟基脯氨酸的氨基酸铜复合物与丙烯酰氯、甲基丙烯酰氯、乙基-丙烯酰氯或丙基-丙烯酰基反应,之后用硫化氢气体流或硫化钠的酸性溶液处理来产生无保护的单体。国际专利申请公开wo2017/055536和实施例中公开了合成方法。
[0111]
在特定的实施方案中,主要单体是丙烯酰胺的衍生物,并且包括二甲基-丙烯酰胺、n-异丁基-丙烯酰胺、n-叔丁基-丙烯酰胺、n-羟乙基-丙烯酰胺、n-(2-羟丙基)-丙烯酰胺、n-(3-羟丙基)-丙烯酰胺、n-(3-羟丙基)-甲基丙烯酰胺、n-(2-羟丙基)-甲基丙烯酰胺、n-(3-氨基丙基)-丙烯酰胺盐酸盐、或n-(3-氨基丙基)-甲基丙烯酰胺盐酸盐。在此,二甲基丙烯酰胺优选地被理解为n,n-二甲基丙烯酰胺。优选地,如本文所理解的主要单体也被称为具有至少一个乙烯基且不含氨基酸部分或叠氮基部分的单体。
[0112]
在其他特定实施方案中,主要单体是丙烯酸的衍生物,包括甲基丙烯酸、丙烯酸2-羟乙酯、丙烯酸2-羟丙酯、丙烯酸3-羟丙酯、丙烯酸2-羟基-1-甲基乙基酯、丙烯酸2-氨基乙酯盐酸盐、甲基丙烯酸3-羟丙酯、甲基丙烯酸2-羟基-1-甲基乙基酯、甲基丙烯酸2-羟乙酯、甲基丙烯酸2-羟丙酯和甲基丙烯酸2-氨基乙酯盐酸盐。优选地,如本文所理解的主要单体也被称为具有至少一个乙烯基且不含氨基酸部分或叠氮基部分的单体。
[0113]
包含一种或多种类型的式i的共-主要单体和任选的式ii和/或式iii的共-主要单体的共聚物通常是以自由基聚合反应制备的。重要的是,本公开的共聚物具有窄尺寸分布,因为在各种疗法中,特别是在癌症疗法中,必须精确地控制载药量。如果不仔细控制,可能会遇到过度投配或投配不足效应。为了获得具有窄尺寸分布的共聚物,必须控制聚合过程中自由基的数量。这可以通过使用包括原子转移自由基聚合(atrp)、氮氧化物介导的聚合(nmp)或可逆加成-片段化-链转移聚合(raft聚合)在内的聚合技术来实现。对于本文所述
的共聚物而言,raft是最优选的技术,因为它与广泛的单体,特别是丙烯酸类相容,并且可以在水性体系中容易地执行。此外,raft聚合可用于嵌段共聚物的合成。另外,raft基团可用于将反应性部分加成至聚合物的头基(例如,用于与抗体或适体缀合)。raft技术是由联邦科学与工业研究组织(commonwealth scientific and industrial research organization,csiro)的研究小组发明的。chiefari等人,(1998)。链尺寸分布的控制是经由从生长中的聚合物链到链转移剂的链转移反应而实现的。所谓的raft试剂形成中间体,并且能够片段化为传播链(称为r基团)和稳定化部分(称为z基团)上的自由基。因此,自由基的数量是有限的,并且所有生长中的聚合物链都具有相似的传播可能性,从而导致共聚物具有窄尺寸分布。在raft聚合中获得的典型的多分散指数(pdi)[定义为mw/mn,其中mw是聚合物的重均摩尔质量,并且mn是聚合物的数均摩尔质量]在1.05至1.4的范围中。合适的raft试剂是硫代羰基巯基化合物。硫代羰基巯基化合物可分为四大类,即二硫代苯甲酸酯、三硫代碳酸酯、二硫代氨基甲酸酯和黄原酸酯。
[0114]
因此,本公开的典型聚合混合物包含主要单体和共-主要单体、raft试剂和自由基引发剂,所述自由基引发剂优选地也称为“用于生成自由基的引发剂体系”。然后将混合物倒入合适的容器中,在所述容器或模具中引发聚合。引发剂可以是热引发剂(例如,va-044),所述热引发剂在高温下失稳以产生反应性自由基;氧化还原引发剂;或光引发剂。用于水溶液中聚合的优选氧化还原引发剂是过氧化物(例如过硫酸铵或过硫酸钾)与硫代硫酸钠或偶氮型化合物(例如2,2'-偶氮双[2-(2-咪唑啉-2-基)丙烷]二盐酸盐或4,4'-偶氮双(4-氰基戊酸))的组合。对于在非水溶剂中的聚合反应,偶氮型引发剂/催化剂,例如,偶氮双(异丁腈)(aibn)、1,1'-偶氮双(环己烷-1-腈)、2,2'-偶氮双(4-甲氧基-2,4-二甲基戊腈)为优选的。还可以使用聚合物改性的偶氮型引发剂,例如(聚二甲基硅氧烷、聚乙二醇)。上述引发剂往往在较高温度下失稳,从而导致形成反应性自由基。
[0115]
另选地,可以在对能够引发乙烯或丙烯酸类单体的聚合的波长的辐射来说透明的容器中使单体发生光聚合。合适的光引发剂化合物可来自i型,例如α-氨基烷基苯甲酮(α-amino alkylphenone);或来自ii型,例如二苯甲酮。也可以使用允许使用较长波长的光敏剂。取决于所用的引发剂化合物,通过加热、辐射或添加催化剂来引发聚合。在本公开的一些实施方案中,有用的是在共聚物(含有主要单体和共-主要单体的混合物)聚合之前合成由亲水性主要单体的10-25个单体单元构成的大分子raft或raft预聚物。由此,可以提高往往为疏水的raft试剂的亲水性,从而促进在水性环境中的聚合反应。
[0116]
在其他实施方案中,通过整合3-25个单元的水溶性单分散聚乙二醇(peg)间隔基来对raft试剂本身进行化学改性。优选地,peg间隔基团在本文中被理解为根据式-(och2ch2)
n-或-(ch2ch2o)
n-的部分,其中n是3至25的整数。经改性的raft试剂表现出改善的水溶性,并且使得能够在一个聚合步骤中合成含亲水氨基酸的共聚物。
[0117]
在其它实施方案中,可能有用的是在聚合物类似反应中生成式i的辅主要结构单元。为此,首先根据上述工序制备raft共聚物,不同之处在于单体混合物仅由主要单体和一种或多种式ii的单体以及任选的一种或多种式iii的单体组成。在纯化该共聚物并去除raft基团后,用含有连接基团和叠氮基的胺反应剂处理该共聚物,从而生成在侧链中具有叠氮基的共聚物。为了清楚起见,该聚合物类似反应的所得共聚物具有与通过包括式i的单体的共聚反应生成的共聚物相同的结构。为了合成具有不同侧链基团的共聚物,例如式i和
ii和/或iii的混合物可以通过改变聚合物类似反应中存在的胺反应性叠氮基的摩尔比来合成,结果是不是共聚物侧链中的所有氨基都被转化。
[0118]
由于已知raft试剂在胺的存在下不稳定,并且是所获得的共聚物的强烈臭味的原因,因此一旦聚合和官能化过程完成,往往应使所述raft试剂失活。在本公开中用于raft基团失活的优选方法是与亲核试剂反应、热消除,或与引发剂和质子给体试剂的组合或过量的官能化引发剂进行第二反应。
[0119]
因为本公开的共聚物旨在用于患者中的药物递送,所以通常优选的是在聚合后或官能化(即,有效负载、靶向部分等的偶联)后纯化共聚物。该步骤去除了潜在有害的成分,包括残留的引发剂、单体或催化剂。用于本发明的共聚物的优选的纯化方法是透析、切向流过滤和毛细管超滤。
[0120]
应当注意的,限定有用的参数值不需要过多的工作,因为参数的数量是有限的,并且一些参数值的优选范围是已知的。含氨基酸的共聚物中的共-主要单体的水平为聚合混合物中存在的所有单体的约2%(mol)与95%(mol)之间,优选地约3%(mol)与40%(mol)之间,最优选地4%(mol)与25%(mol)之间。含氨基酸的共聚物(不含细胞类型或组织类型特异性靶向部分)的平均分子量通常在约5,000道尔顿与80,000道尔顿之间,优选地在约5,000da与40,000da之间,最优选地在约5,000da与20,000da之间。本公开的共聚物可含有的共-主要单体的数量已经在上面针对三种情况进行了讨论,所述三种情况即,针对含有式i的单体作为唯一类型单体的共聚物;针对含有式i的单体以及式ii和/或iii的单体,由此在聚合之后只有式i的单体被官能化(偶联至第一有效负载)的共聚物;以及针对其中所有共-主要单体被官能化(偶联至第一有效负载和第二有效负载)的类似共聚物。
[0121]
如本文所理解的,与特定类型的单体相关的术语“单体水平”优选地被定义为由于特定类型单体导致的重复单元数与由于(共)聚合物分子中的所有单体导致的重复单元数之比。如本文所理解的水平优选以百分比值表示。
[0122]
一旦共聚已经完成,本发明的包含式i的共-主要单体的共聚物就准备好用活性剂分子和/或细胞类型特异性或组织类型特异性靶向部分(例如,抗体)进行官能化。这种官能化导致在共聚物与活性剂分子和/或靶向部分之间建立共价键。在某些活性剂(例如某些放射性同位素)的情况下,将共聚物用螯合剂官能化,并且活性剂由所述螯合剂保持。需注意,取决于要接合到共聚物的特定活性剂的性质,在引入活性剂之前,共聚物可以用细胞类型特异性或组织类型特异性靶向部分官能化,或者活性剂可以在用靶向部分官能化之前接合至共聚物。
[0123]
式i的共-主要单体含有叠氮基部分。叠氮基部分是一种高反应性实体,其用于与炔烃进行的所谓“点击反应(click reaction)”。与胺或硫醇加成反应相比,叠氮基-炔烃或四嗪-反式环辛烯点击反应是快速和高选择性的。事实上,由于生物环境(肽、蛋白质)中氨基和硫醇基团的存在,加成反应不会遇到副反应,从而由于叠氮基的正交反应性而使所述叠氮基成为用于药物递送应用的完美选择。此外,含有侧链连接的氨基酸的单体和包含适当比例的此类单体(至少2mol%)的(共)聚合物具有低毒性的优点。例如,丙烯酰基-赖氨酸单体和含有这种单体的(共)聚合物在浓度为至多5mm的细胞培养实验中不显示任何毒性作用。因此,理想的药物载体可以通过用叠氮基官能化丙烯酰基-赖氨酸或其它侧链连接的氨基酸,从而提供具有低毒性潜力的生物相容性共聚物来设计。在抗体药物缀合物(adc)领域
中,经常使用高毒性药物,从而使得此类adc的制备昂贵且从安全角度来看具有挑战性。在本公开中,引入了侧反应性载体共聚物,所述侧反应性载体共聚物可以与抗体偶联以生成“药物反应性”抗体聚合物缀合物。细胞毒性有效负载(即,细胞毒性药物或保持细胞毒性药物的螯合剂)在生产的最后阶段与缀合物偶联。因此,可以减少其中存在高效力(high potency,hipo)物质的生产步骤和下游工艺的数量。此外,本文提出的技术提供了高度的灵活性,因为有效负载也可以在抗体附着之前偶联到聚合物载体。这对于对降解敏感的抗体或其他癌细胞特异性靶向部分尤其有用。
[0124]
本公开的共聚物还可包含式ii和/或iii的共-主要单体。后一种共-主要单体含有侧链连接的氨基酸,所述侧链连接的氨基酸含有游离的或官能化的α-氨基和/或羧基。其中α-氨基或羧基中的至少一者未被官能化的包含式i的含叠氮基的单体和式ii和/或iii的单体的共聚物,使得抗体与两种不同的有效负载分子能够“一锅”偶联。例如,将第一有效负载分子(例如针对放射性同位素的螯合剂)用叠氮基反应性部分(例如,应变炔烃)官能化,并将第二有效负载分子(例如细胞毒性药物)用胺反应性基团(例如,nhs酯)官能化。这使得药物载体能够用于强效的联合疗法,而没有副反应和“错误标记”的风险,因为两个反应是正交的。此外,可以减少反应和纯化步骤(例如透析)的数量,这可用于敏感和高效的有效负载分子。此外,在癌症疗法应用中,本文提出的组合方法使得共聚物能够装载细胞毒性剂和诊断性放射性同位素,并且因此是可追踪的。将细胞毒性剂递送到甚至很小的肿瘤转移处之后,可以进行高分辨率的正电子放射断层造影术(positron emission tomography,pet),从而提供关于疗法进展的有用信息。
[0125]
抗体药物或聚合物缀合物中癌细胞特异性靶向部分的亲水/疏水平衡对于构建体的保质期和循环时间至关重要,因为聚集倾向于缩短保质期和循环时间两者,从而导致降低的疗法功效。由于药物连接至载体而不是直接连接至抗体,因此聚合物药物载体的使用有望降低由高疏水性有效负载(例如某些抗癌药物)引起的聚集风险。然而,这种积极效应可能会被通过使用药物载体所带来的高得多的载药量所削弱或甚至抵消。在本公开的共聚物的一些实施方案中,后一个问题通过在共聚物中包含式ii和/或iii的未官能化的共-主要单体来解决。因此,使用叠氮基官能化的侧链连接的氨基酸(例如丙烯酰基-赖氨酸-叠氮化物)来将有效负载偶联到载体共聚物,并且高度亲水的未修饰的侧链连接的氨基酸(例如丙烯酰基-赖氨酸)使得能够提高疏水性有效负载的“溶解性”。装载有疏水性有效负载的聚合物载体的总体“疏水性”降低,并随之降低了聚集的可能性。
[0126]
在具体实施方案中,活性剂(在此处是在癌症疗法中使用的细胞毒性药物或分子)可以是微管抑制剂,例如单甲基奥瑞斯他汀e(mmae)或emtansine(dm1);插入药物,例如多柔比星;烷基化剂,例如环磷酰胺(cp);抗代谢物,例如5-氟尿嘧啶(5-fu);激素或激素受体调节剂,例如枸椽酸他莫昔芬;酪氨酸激酶抑制剂,例如阿法替尼或博舒替尼;基于肽的毒素,例如α-鹅膏蕈碱;免疫检查点抑制剂,例如或适用于抗体指导的酶前药疗法(adept)的酶;,能够干扰一种或多种基因或其相应信使rna、sirna、微rna或反义rna的基于多核苷酸的药物;或放射性同位素,例如但不限于氟-18、铜-64、镓-68、锆-89、铟-111、碘-123(诊断应用)或铜-67、锶-89、钇-90、碘-131、钐-153、镥-177、镭-223和锕-225(治疗应用)。
[0127]
在特定实施方案中,活性剂(在此为在癌症疗法中使用的细胞毒性药物或分子)可
以是放射性同位素/放射性核素,例如但不限于氟-18、钪-43、钪-44、铜-61、铜-64、镓-68、锆-89、铟-111、碘-123、铽-152、铽-155(诊断应用)或钪-47、铜-67、锶-89、钇-90、碘-131、铽-149、钐-153、铽-161、镥-177、镭-223和锕-225(治疗应用)。
[0128]
本发明人已经惊奇地发现,根据本发明的包含第一有效负载分子和第二有效负载分子的共聚物,其中第一有效负载分子和第二有效负载分子是可用于联合疗法的活性剂,可用于将这两种活性剂共递送至同一癌细胞。细胞毒性有效负载可以优选地是除了其细胞毒性潜力之外,还(例如通过抑制dna修复机制(例如蛋白激酶抑制剂)或通过直接靶向dna菌株(例如,如在多柔比星的情况下通过嵌入))影响癌细胞的“放射敏感性”的试剂。这种试剂在本文中优选称为“放射增敏剂”。在这种情况下,此类“放射增敏剂”可在与放射性核素的联合治疗中使用,因为放射疗法的细胞杀伤效应得到了增强。因此,本文公开的载体技术具有以下优点:两种试剂可以结合到相同的载体分子并偶联相同的癌细胞特异性靶向部分,以确保“共递送”到相同的癌细胞。例如,将第一有效负载分子(例如针对放射性同位素的螯合剂)用叠氮基反应性部分(例如,应变炔烃)官能化,并将第二有效负载分子(例如细胞毒性药物)用胺反应性基团(例如,nhs酯)官能化。
[0129]
优选地,如本文所理解的用于与放射性同位素联合应用的放射增敏剂是激酶抑制剂,优选地选自阿利塞替布(alisertib)、mk-1775、mk-2206、赛卡替尼(saracatinib)和替西罗莫司(temsirolimus),更优选地选自赛卡替尼和mk-2206。优选地,如本文所理解的用于与放射增敏剂联合应用的放射性同位素选自镥-177和铽-161。因此,优选地,在本发明的范围内,用于联合应用的放射增敏剂和放射性同位素选自阿利塞替布和镥-177、阿利塞替布和铽-161、mk-2206和镥-177、以及mk-2206和铽-161。
[0130]
如本文所理解的,阿利塞替布是根据下式的化合物:
[0131][0132]
其可以被包含在本发明的共聚物中,例如通过经由由其羧基形成的肽键附接。
[0133]
如本文所理解的,mk-2206是根据下式的化合物:
[0134][0135]
其可以被包含在本发明的共聚物中,例如通过经由由其氨基形成的肽键附接。本发明人还已惊奇地发现,本发明的共聚物适用于诊断应用,以及组合的诊断和治疗应用。组合的诊断和治疗应用也可以被称为治疗诊断应用。优选地,根据本发明,本发明的共聚物可包含细胞类型特异性或组织特异性靶向部分,例如靶向特定类型癌细胞的抗体,以及作为具有放射性核素的螯合剂的有效负载分子。如本文所公开的某些放射性核素可以被监测,例如铽161由于其γ-发射而可以用γ相机可视化,并且因此可用于检测抗体所靶向的癌组织或细胞类型。可用于靶向α疗法的铽-149,在pet扫描中具有可见性并且因此可以被监测。根据本公开,氟-18、钪-43、钪-44、铜-61、铜-64、镓-68、锆-89、铟-111、碘-123、铽-152、铽-155在如本文所述的诊断应用中特别有用,并且可被称为在诊断中可用的放射性核素。正如本领域技术人员所知,可通过使用合适的方法来监测诊断中可用的放射性核素,所述合适的方法为例如闪烁扫描术、单光子发射计算机断层扫描术(single photon emission computed tomography,spe-ct);或正电子发射断层摄影计算机断层扫描术(positron emission thomography computed tomography,pet-ct)。根据本发明的共聚物的此类用途优选允许监测共聚物的生物分布。共聚物的生物分布在本文中被理解为在投配所述共聚物后在受试者,优选患者的组织内的分布。本领域技术人员将理解,其中活性剂包含可用于治疗应用的放射性核素,例如选自铜-67、锶-89、钇-90、碘-131、钐-153、镥-177、镭-223和锕225的放射性核素(这些放射性核素可称为可用于疗法中的放射性核素)的此类共聚物将具有与其中放射性核素可用于诊断应用的共聚物基本上相同的生物分布。因此,根据本发明,本发明的共聚物可优选用于在疗法期间监测治疗性共聚物的生物分布。例如,为此目的,包含作为可用于疗法的放射性核素的活性剂的共聚物可以优选用小于10重量%的如本文所定义的其中有效负载包含可用于诊断的放射性核素的共聚物来补充。进一步优选地,用于在组合治疗和诊断应用中使用的本发明的共聚物可包含两种放射性核素,一种放射性核素可用于疗法中并且另一种放射性核素可用于诊断,例如分别被包含在第一有效负载分子和第二有效负载分子中。优选的是其中可用于疗法的放射性核素和可用于诊断的放射性核素是相同元素的同位素的组合。因此,优选的组合包含钪-43和钪-47、铜-61和铜-67、铜-64和铜-67、碘-123和碘-131、铽-152和铽-161、以及铽-155和铽-161。进一步优选的组合包括两种不同元素的同位素,例如铟-111和镥-177,以及铟-111和铽-161。本发明的共聚物也可用于诊断监测。例如,包含靶向特定癌组织类型的靶向部分的共聚物的生物分布的变化可以指示靶向所述癌组织的疗法的进展,所述共聚物还包含用于诊断的放射性核素。特别地,这种方法可以可用于癌症监测,优选地监测已经转移的癌症的疗法。如本文所公开的,本发明的共聚物可能可用于癌症监测,在本文中被定义为受试者,优选患者体内癌症组织的分布。
[0136]
在其他特定实施方案中,活性剂是细胞毒性药物和能够例如通过抑制抗凋亡因子
(例如bcl-2)或靶向细胞外排泵(例如mdr-1转运体)来克服肿瘤细胞抗性的药物的组合。
[0137]
前述活性剂是与本公开的共聚物相容的试剂和试剂类别的非限制性示例,并且在不超出本公开的范围的情况下,本领域技术人员可以使用所公开的试剂和试剂类别的变体或衍生物。
[0138]
取决于活性剂或其它有效负载分子的结构,所述活性剂或其它有效负载分子可以直接偶联到共聚物中含有的式i的共-主要单体的叠氮基部分或者式ii或iii的共-主要单体的α-氨基或α-羧基,或者所述活性剂或其它有效负载分子可以经由连接基团结构偶联至所述共聚物。此类连接基团可用作活性剂与共聚物之间的简单间隔基,用作共聚物药代动力学的改性剂,或者含有使得能够进行或促进活性剂在靶细胞中释放的元件。连接基团应在存储期间和稍后在血液中为稳定的,以避免意外释放活性剂。从共聚物中释放活性剂应仅在靶细胞内部发生。因此,有用的连接基团(专注于癌症疗法)应对细胞间因素敏感,所述细胞间因素为例如半胱天冬酶或组织蛋白酶、葡糖醛酸酶(gusb)(基于β-葡糖苷酸的连接基团)、酸性ph(在肿瘤组织或细胞器[溶酶体]中存在)、或还原环境(响应于细胞间谷胱甘肽浓度增加)。另一可能性将是使用二胺型或硫醚型的不可降解连接基团,所述不可降解连接基团不是特定酶的靶标,并且仅在具有溶酶体或过氧化物酶体的苛刻条件下降解。后一种连接基团类型是优选的,因为它与最大的血清稳定性和降低的非特异性毒性相关联。
[0139]
本公开的共聚物通常用细胞类型或组织类型特异性靶向部分官能化。虽然这种官能化步骤可以在活性剂已经偶联到共聚物之后执行,但是往往有利的是(特别是当使用高细胞毒性剂或半衰期短的放射性同位素时)首先制备共聚物和靶向部分的缀合物。然后,共聚物的活性剂负载可在向受试者施用之前不久发生。潜在的靶向部分为但不限于单克隆抗体,包括免疫检查点抑制剂、抗体片段、纳米抗体(单结构域抗体)、darpin、肽激素、能够与细胞表面受体结合的非抗体蛋白、基于dna/rna的适体以及能够与细胞表面受体(例如,在肿瘤环境中的叶酸或生物素)结合的小分子。在癌症疗法的情况下,有利的是如果上述靶部分在健康组织中具有低到可忽略的表达水平并且在癌细胞的细胞表面上具有高表达水平/拷贝数,以避免副作用。潜在的靶标是但不限于cd19(b淋巴细胞表面抗原b4)、cd20(b淋巴细胞抗原)、cd21(补体受体2型,cr2)、cd22(分化簇-22)、cd40(分化簇-40)、cd52(campath-1抗原)、cd152(etla-4,细胞毒性t淋巴细胞相关蛋白4)、cd180(rp105)、cd274(pd-l1,程序性细胞死亡1配体1)、cd279(pd-1,程序性细胞死亡蛋白1)、egfr(表皮生长因子受体)、fap(成纤维细胞激活蛋白)、gd2(二唾液酸神经节苷脂)、gitr(糖皮质激素诱导的tnfr家族相关基因)、her2(人表皮生长因子受体2,erbb2,erb-b2受体酪氨酸激酶)、kir2dl1(杀伤细胞免疫球蛋白样受体2dl1)、nkg2d(klrk1)、msln(间皮素)、pdgf(血小板衍化生长因子)、pdgfr(血小板衍化生长因子受体)、vegf(血管内皮生长因子)、vegfr(血管内皮生长因子受体)、cae(癌胚抗原)、ca9/ca ix(碳酸酐酶ix)、α-叶酸受体(叶酸受体1)、和psma(前列腺特异性膜抗原)。
[0140]
靶向部分与共聚物的共价附接应以位点特异性方式进行,以获得均质产物,以及保持靶向部分的结合亲和力。合适的偶联策略是与肽标签进行酶催化反应(例如分选酶介导的偶联)、醛标签或转谷氨酰胺酶标签,或共聚物与靶向部分之间的所谓“点击”反应。后一过程可以通过在将反应性、非规范(非天然)氨基酸合成为蛋白质靶向部分(例如,抗体)期间经由整合来实现(例如,借助于使用由非天然氨基酸的trna识别的重编程终止密码子
的密码子扩展技术)。
[0141]
分选酶是指通过识别和分裂羧基末端分选信号来修饰表面蛋白的一组原核酶。对于金黄色酿脓葡萄球菌(staphylococcus aureus)来源的酶,识别信号由基序lpxtg(leu-pro-any-thr-gly)组成,并且对于酿脓葡萄球菌(staphylococcus pyogenes)来源的酶,识别信号是lpxta(leu-pro-any-thr-ala)。信号序列之前是高度疏水的跨膜序列和一系列碱性残基(例如精氨酸)。分裂发生在信号序列的thr和gly/ala残基之间,thr残基瞬时附接至分选酶的活性位点cys残基,之后进行转肽作用,所述转肽作用使蛋白质共价附接至细胞壁组分(例如,革兰氏阳性细菌的肽聚糖层)。cozzi,r.等人,(2011)faseb j 25(6):1874-86。该酶促机制可适于实现肽或蛋白质的融合,并且最近已用于制备adc。欧洲专利申请号20130159 484(ep 2 777 714);beerli,rr等人,(2015)plos one 10(7):e0131177。在所公开的方法中,对单克隆抗体进行遗传修饰以在其重链和轻链的c末端处含有分选酶基序,并且将细胞毒性药物修饰为含有寡聚甘氨酸段。分选酶催化的反应高效地将经修饰的药物分子添加到抗体链的c末端,从而产生了均质的adc。
[0142]
通过用寡聚甘氨酸段修饰本公开的共聚物的头基,所述共聚物本身变成了分选酶催化反应的靶标。由于共聚物可负载有多种活性剂,因此这种方法产生了这样的adc,所述adc中的许多活性剂分子与抗体中的少量限定(无害)位点连接(每个抗体分子2-4个c末端分选酶标签)。因此,dar升高,并且adc的效力随之提高。可以在聚合开始时使用最近开发的含有2-8个甘氨酸残基的raft试剂引入共聚物的寡聚甘氨酸段。当使用该官能化的raft试剂时,在每个共聚物分子中仅存在一个分选酶基序。
[0143]
另一种酶促偶联方法利用转谷氨酰胺酶催化反应。转谷氨酰胺酶,也称为蛋白质-谷氨酰胺γ-谷氨酰转移酶,通常通过将一种蛋白质的谷氨酰胺残基的γ-羧基酰胺基转移至同一或另一蛋白质的赖氨酸残基的ε-氨基上来使蛋白质交联。在过去的二十年中,这些酶被用于各种领域,如食品工业如“肉胶”(martins im等人,(2014),appl.microbiol.biotechnol.98:6957-64?)、组织工程化(ehrbar m.等人,(2007)bio-macromolecules,8(10):3000-7)、治疗性蛋白质的修饰(mero a.等人,(2011)j control release,154(1):27-34)或基因递送(trentin d.等人,(2005)j control release,102(1):263-75)中。
[0144]
在这种情况下,微生物转谷氨酰胺酶(mtg)是优选类别的酶,因为它们是与内源性人类转谷氨酰胺酶相比,与钙和核苷酸无关的酶。与人类转谷氨酰胺酶的四个结构域相比,它们由单个结构域组成,并且分子量为人转谷氨酰胺酶的约一半。此外,mtg在较大的ph值、缓冲液和温度范围内运行,并且具有大量潜在的底物。kieliszek m等人,(2014)rev folia microbiol.59:241-50;martins im.等人,(2014)。
[0145]
与分选酶介导的偶联策略类似,通过修饰raft试剂将转谷氨酰胺酶基序引入本公开的共聚物的头基,从而确保每条聚合物链仅引入一个转谷氨酰胺酶基序。适当的基序是小肽,例如但不限于作为潜在赖氨酸供体序列的fkgg(ehrbar m.等人,(2007)),以及作为谷氨酰胺受体序列的lqsp或tqga(caporale a.等人,(2015)biotechnol j.10(1):154-61)[在这种情况下,使用癌细胞特异性靶向部分中的反应性赖氨酸残基];或作为潜在谷氨酰胺受体序列的3-25个单位长度的含有末端氨基的单分散peg间隔基。
[0146]
该策略的一种变体利用转谷氨酰胺酶将点击反应基团(例如叠氮化物或四嗪)定
点附接到靶向部分(例如单克隆抗体),该抗体连接的反应基团随后用于与在本公开的共聚物的聚合头基处的“相对”点击反应性基团(炔烃或/张力烯烃)反应。在共聚物/抗体处提到的反应性部分是指为可互换的。这种优选的策略在本文给出的实施例(实施例16、实施例23和实施例34)中使用。
[0147]
可以采用其他用于将靶向部分连接到共聚物的方法。靶向抗体或其他多肽可以例如通过将氨基酸侧链中的羟基官能团转化为反应性醛而在翻译后改变。在基于多核苷酸的靶向部分(例如适体)的情况下,可以通过在固相合成期间与整合到适体中的反应性官能团(例如胺、硫醇、醛)反应来实现与本公开的共聚物的偶联。可以使用本领域中众所周知的其他定点偶联技术将共聚物偶联至靶向部分。
[0148]
药物组合物
[0149]
本公开的药物组合物包含有效量的本公开的活性部分,所述活性部分是与一种或多种药学上可接受的载体或赋形剂一起配制的。
[0150]
本公开的药物组合物可以肠胃外地施用,通过吸入喷雾、眼中局部地、经直肠地、经鼻地、经颊地、经阴道地或经由植入的储库来施用,优选地通过注射(或输注)施用。本公开的药物组合物可含有任何常规无毒的药学上可接受的载体、佐剂或媒介物。在一些情况下,可以用药学上可接受的酸、碱或缓冲液调节制剂的ph,以增强所配制的活性部分或其递送形式的稳定性。如本文所用的术语肠胃外包括皮下、皮内、静脉内、肌内、关节内、动脉内、滑膜内、胸骨内、鞘内、病灶内和颅内注射或输注技术。
[0151]
可以根据已知技术,使用合适的分散剂或润湿剂和悬浮剂来配制可注射制剂,例如无菌可注射水性或油质悬浮液。无菌可注射制剂也可以是在无毒的肠胃外可接受的稀释剂或溶剂中的无菌可注射溶液、悬浮液或乳液。可以采用的可接受的媒介物和溶剂是水、林格氏溶液、u.s.p.和等渗氯化钠溶液。增溶赋形剂包括水溶性有机溶剂,例如聚乙二醇300、聚乙二醇400、乙醇、丙二醇、甘油、n-甲基-2-吡咯烷酮、二甲基乙酰胺和二甲基亚砜;非离子表面活性剂,例如cremophor el、cremophor rh40、cremophor rh60、solutol hs15、d-α-生育酚聚乙二醇1000琥珀酸酯、聚山梨醇酯20、聚山梨醇酯80、脱水山梨糖醇单油酸酯、泊洛沙姆407、labrafil m-1944cs、labrafil m-2125cs、labrasol、gellucire 44/14、softigen 767,以及peg 300、peg 400和peg 1750的单脂肪酸酯和二脂肪酸酯;水不溶性脂质,例如蓖麻油、玉米油、棉籽油、橄榄油、花生油、薄荷油、红花油、芝麻油、大豆油、氢化植物油、氢化大豆油,以及椰子油和棕榈籽油的中链甘油三酸酯;各种环糊精,例如α-环糊精、β-环糊精、羟丙基-β-环糊精(例如,kleptose)和磺丁基醚-β-环糊精(例如,captisol);以及磷脂,例如卵磷脂、氢化大豆磷脂酰胆碱、二硬脂酰磷脂酰甘油、l-α-二肉豆蔻酰磷脂酰胆碱和l-α-二肉豆蔻酰基-磷脂酰甘油。strickley(2004)pharm.res.21:201-30。
[0152]
可注射制剂可被灭菌,例如,通过滤过保留细菌的过滤器,或通过在无菌固体组合物中掺入灭菌剂(或通过辐射对固体组合物进行灭菌),随后可在使用前将所述经灭菌的可注射制剂溶解或分散在无菌水或其他无菌可注射介质中。
[0153]
为了延长活性剂的效应,往往期望减慢来自皮下或肌内注射的活性剂的吸收。肠胃外施用的活性部分的延迟吸收是通过将活性部分溶解或悬浮在油媒介物中而实现的。通过将活性部分微囊包封在可生物降解的聚合物(例如聚丙交酯-聚乙交酯)中来制备可注射的贮库形式。取决于活性部分与聚合物的比率和所采用的特定聚合物的性质,可以控制活
性剂的释放速率。其他可生物降解的聚合物的示例包括聚(原酸酯)和聚(酸酐)。还可以通过将活性部分截留在与身体组织相容的脂质体或微型乳剂中来制备贮库型可注射制剂。
[0154]
用于直肠或阴道施用的组合物优选地为栓剂,所述栓剂可以通过将本公开的活性部分与合适的无刺激赋形剂或载体(例如可可脂、聚乙二醇或栓剂蜡)混合而制备,所述赋形剂/载体在环境温度下为固体,但在体温下呈液态,因此在直肠或阴道腔内融化并释放出活性部分(并且因此,释放出活性剂)。
[0155]
眼科制剂、滴耳剂、眼药膏、粉剂和溶液剂也被认为在本公开的范围内。
[0156]
对于肺部递送,本公开的药物组合物被配制,并通过直接施用(例如吸入到呼吸系统中)而以固体或液体微粒形式施用给患者。为实践本公开而制备的活性部分的固体或液体微粒形式包括可呼吸大小的粒子:即,大小小到足以在吸入时穿过口和喉并进入支气管和肺的肺泡的粒子。气雾化的治疗剂,尤其是气雾化的抗生素的递送在本领域中是已知的(参见例如美国专利号5,767,068、美国专利号5,508,269和wo 98/43650)。在美国专利号6,014,969中也找到了关于抗生素的肺部递送的讨论。
[0157]
以单剂量或分剂量施用给人类受试者或患者的本公的活性部分的总每日剂量优选地包括0.01mg/kg体重至50mg/kg体重的活性剂,或更优选地0.1mg/kg体重至30mg/kg体重的活性剂。单剂量组合物可含有此类量或其约数以组成每日剂量。一般而言,根据本公开的治疗方案包括每天以单剂量或分剂量向需要此类治疗的人类受试者施用约1mg至约5000mg的活性剂(包含在本公开的活性部分中)。用于哺乳动物的剂量可以根据后一人类剂量来估算。
[0158]
对于包含放射性核素,特别是可用于疗法的放射性核素的本发明共聚物,放射性核素的剂量也可以以放射性单位描述,优选以mbq/kg体重描述。以单剂量或分剂量施用于人类受试者或患者的包含本发明的放射性核素的共聚物的总日剂量优选在3mbq/kg体重与300mbq/kg体重之间。如技术人员所知,进一步优选的投配方案取决于所用的放射性核素。对于包含钇-90的本发明共聚物,以单剂量或分剂量施用于人类受试者或患者的总日剂量优选在5mbq/kg体重至35mbq/kg体重之间,甚至更优选地在7mbq/kg体重与25mbq/kg体重之间,最优选地在10mbq/kg体重与15mbq/kg体重之间。对于包含镥-177的本发明共聚物,以单剂量或分剂量施用于人类受试者或患者的总日剂量优选在5mbq/kg体重与100mbq/kg体重之间,甚至更优选地在10mbq/kg体重与80mbq/kg体重之间,最优选地在10mbq/kg体重与60mbq/kg体重之间。如本文所理解的,单剂量组合物可含有此类量或其约数以组成每日剂量。如本文所理解的,技术人员将能够取决于放射性核素且取决于期望的应用(例如实体瘤的治疗、血液肿瘤的治疗、淋巴细胞去除以实现有效的cart-t治疗)来确定优选的剂量。
[0159]
本公开的活性部分可以例如通过静脉内、动脉内、真皮下(subdermally)、腹膜内、肌内或皮下(subcutaneously)注射;或经颊地、经鼻地、经粘膜地、局部地、在眼用制剂中、或通过吸入,作为包含约0.01mg/kg体重至约50mg/kg体重的活性剂的每日剂量来施用。另选地,可以每4至120小时或根据特定活性部分的要求来施用剂量(基于后一种活性剂每日剂量)。本文的方法设想施用有效量的活性部分(在药物组合物中)以实现期望或所述的效应。通常,本公开的药物组合物将每天施用约1次至约6次,或者另选地作为连续输注施用。此类施用可以用作慢性或急性疗法。可以与药学上可接受的赋形剂或载体组合以产生单一剂型的活性部分的量将取决于所治疗的宿主和特定的施用模式而变化。典型的组合物将含
有约5%至约95%的活性部分(w/w)。另选地,此类制剂可含有约20%至约80%活性的活性部分。任何特定患者的具体剂量和治疗方案将取决于多种因素,包括所采用的特定活性部分的活性;年龄;体重;总体健康状况;性别;饮食;施用时间;排泄速率;药物组合;疾病、病症或症状的严重程度和病程;患者对疾病、病症或症状的倾向;以及治疗医师的判断。
[0160]
本技术中引用的所有参考文献,包括出版物、专利和专利申请,均应被视为已整体并入。
[0161]
除非本文另有指示,否则本文中对值范围的描述仅旨在用作引用落入该范围的每个单独值的速记方法,并且每个单独值并入本说明书中,如同其在本文中被单独引用一样。除非另有说明,否则本文提供的所有精确值都代表对应的近似值(例如,针对特定因子或测量值提供的所有精确示例性值可被为还提供了对应的近似测量值,在适当情况下用“约”修饰)。
[0162]
除非另有说明或明显与上下文相矛盾,否则本文中使用例如关于一个或多个要素的术语对本发明的任何方面或实施方案的描述旨在为本公开的“由一个或多个特定要素组成”、“基本上由一个或多个特定要素组成”或“基本上包含一个或多个特定要素”的类似方面或实施方案提供支持(例如,除非另有说明或明显与上下文相矛盾,否则本文描述为包含特定要素的组合物应理解为还描述由该要素组成的组合物)。
[0163]
本发明包括在适用法律所允许的最大范围内本文提出的方面或权利要求中所述主题的所有修改和等同物。
[0164]
通过参考以下实施例,将更容易地理解如此一般描述的本公开,这些实施例是通过举例说明的方式提供的,并且并非旨在限制本发明。
[0165]
实施例
[0166]
注意:在与合成侧链连接的氨基酸有关的实施例中,首先以iupac命名法给出了名称。此后,使用缩写名称。表1显示了对应关系。
[0167]
表1:iupac名称和缩写
[0168]
[0169]
[0170]
[0171][0172]
需注意,本公开中的共聚物是通过受控自由基聚合技术如raft聚合来聚合的,并且具有通常在1.1-1.4范围内的低聚分散指数(pdi)。在本公开中针对例示性聚合物药物载体给出的数字表示共聚物链的平均单体组成。除非另有说明,否则本公开中的共聚物是无规共聚物。
[0173]
实施例1:6-丙烯酰胺基-2-(2-叠氮乙酰胺基)己酸的合成
[0174][0175]
将2-叠氮基乙酸2,5-二氧吡咯烷-1-酯(4.0当量)的四氢呋喃溶液滴加到6-丙烯酰氨基-2-氨基己酸(1.0当量)和碳酸氢钠(2.0当量)在h2o的冷却溶液中。将溶液于0℃搅拌1小时,然后在室温下搅拌过夜。将thf从溶液中减压去除。然后用6m hcl将溶液酸化至ph为1,并用乙酸乙酯萃取(3次)。将合并的有机相经na2so4干燥,并减压浓缩。将所得油状膜溶于少量etoac中,并滴加到庚烷中。滤出细白色沉淀粉末并在真空下干燥,从而得到所需产物(95%产率)。通过nmr光谱验证所获得的化合物的结构。
[0176]
实施例2:6-丙烯酰胺基-2-(6-叠氮己酰胺基)己酸的合成
[0177][0178]
如实施例1所述制备6-丙烯酰胺基-2-(6-叠氮基己酰胺基)己酸(85%产率),区别在于使用6-叠氮基己酸2,5-二氧吡咯烷-1-酯(sigma-aldrich,switzerland,4.0当量)。通过nmr光谱验证所获得的化合物的结构。
[0179]
实施例3:ak-peg
(4)-叠氮化合物的合成
[0180][0181]
如实施例1所述制备ak-peg
4-叠氮化物(82%产率),区别在于使用叠氮基-peg
(4)-nhs(click chemistry tools,scottsdale,usa,4.0当量)。通过nmr光谱验证所获得的化合物的结构。
[0182]
实施例4:经由铜络合物合成(s)-6-丙烯酰胺基-2-氨基己酸单体
[0183]
将l-赖氨酸(14.62g;100mmol)溶于150ml去离子水中,加热至约80℃。在30分钟内分批添加碳酸铜(16.6g;75mmol)。将反应搅拌另外30分钟。将热的深蓝色悬浮液滤过硅胶。将过滤器用少量水洗涤。在第二天,将含有赖氨酸铜复合物的合并滤液在冰浴中冷却,并添加100ml四氢呋喃(thf)。在1小时的时间段内滴加丙烯酰氯在甲基叔丁基醚(tbme)中的溶液(8.9ml,110mmol)。通过平行滴加10%的氢氧化钠溶液将ph最初保持在介于8与10之间。添加一半的丙烯酰氯溶液后,产物开始沉淀。当已添加大部分的丙烯酰氯时,减慢氢氧化钠的添加,以使ph降至约6,并使反应混合物的温度达到室温。将蓝色悬浮液搅拌另外2小时,然后过滤。用水和丙酮洗涤保留在过滤器上的固体物质,然后干燥。获得了产量为6.5g的丙烯酰基-l-赖氨酸铜复合物。
[0184]
将丙烯酰-l-赖氨酸铜复合物(29.5g)悬浮在300ml去离子水中,并在冰浴中冷却。将h2s气体鼓入悬浮液,直到硫化铜沉淀完全。将三克活性炭添加入悬浮液中。将该悬浮液短暂加热至100℃。在冷却至室温后,向悬浮液中添加500ml丙酮,然后将该悬浮液在硅胶上过滤。将澄清的滤液放入旋转蒸发仪中。在蒸发溶剂后,将固体产物从200ml的50%丙酮水溶液中重结晶。得到了产量为17.76g(70%)的白色粉末。通过nmr和lc-ms波谱法验证该化合物的结构。
[0185]
实施例5:合成(2s)-3-(丙烯酰氧基)-2-氨基丙酸
[0186]
将l-丝氨酸(5g,47.6mmol)在水(50ml)中的溶液加热至80℃,并添加固体碳酸铜(5.79g,26.2mmol)。将该溶液搅拌10分钟。随后通过过滤收集未溶解的残余物,并用水(30ml)洗涤。将合并的滤液在冰浴中冷却,并缓慢添加koh(27.1ml,47.6mmol)。向该溶液中滴加丙烯酰氯(4.52ml,59.5mmol)在丙酮(30ml)中的混合物。然后在搅拌下将反应混合物在4℃下孵育过夜。分离出形成的固体,并用水(50ml)/甲醇(50ml)/乙基-叔丁基醚(550ml)(mtbe)洗涤,最后减压干燥,得到o-丙烯酰基-l-丝氨酸-cu
2
复合物(3.8g,10.01mmol;42.1%产率)。随后通过与实施例1中所述类似的工序去除复合物中的铜。得到了产量为1.43g(45%)的丙烯酰基-l-丝氨酸白色粉末。通过nmr波谱法和lc-ms波谱法验证化合物的身份。
[0187]
实施例6:合成(2s)-3-(丙烯酰氧基)-2-氨基丁酸
[0188]
在冰浴中冷却具有6ml三氟乙酸(tfa)的反应容器。随后,添加固体l-苏氨酸(2.00g,16.79mmol),并将混合物搅拌5分钟。添加三氟甲磺酸(0.18ml,2.0mmol),然后添加丙烯酰氯(2.5ml,32.9mmol),并将反应混合物在室温下孵育2小时。在反应完成后,将产物用甲基叔丁基醚(mtbe)沉淀。在分离出固体后,将产物用mtbe和丙酮洗涤。最后,将o-丙烯酰基-l-苏氨酸盐酸盐减压干燥,以得到白色粉末(产率:32%)。通过nmr和lc-ms波谱法验证该化合物的结构。
[0189]
实施例7:合成(s)-3-(4-(丙烯酰氧基)苯基)-2-氨基丙酸
[0190]
按照实施例1中所述的工序执行o-丙烯酰基-l-酪氨酸-cu
2
复合物的合成。通过以下工序从复合物中去除铜:在研磨盘中将73.15g(140mmol)的o-丙烯酰基-l-酪氨酸-cu
2
复合物溶解在220ml 2n hcl中。使用pt 3000设备将混合物均质化。随后,将混合物过滤并将残余物用50ml 2n hcl洗涤两次。然后将固体化合物在40℃下在naoh上减压干燥,得到o-丙烯酰基-l-酪氨酸盐酸盐(46.96g,产率63%)。
[0191]
实施例8:合成(s)-2-(4-丙烯酰胺基苯基)-2-氨基乙酸
[0192]
将boc-4-氨基-l-苯丙氨酸(2.50g,8.9mmol,anaspec,fremont,ca)溶于25ml三氯甲烷中。向该溶液中添加三乙胺(2.47ml,17.8mmol),并将混合物冷却至-15℃。随后,在搅拌下将丙烯酰氯(0.79ml,9.8mmol)在氯仿中的溶液滴加到该混合物中。在丙烯酰氯添加完成后,将反应混合物搅拌另外三小时。然后使反应混合物穿过玻璃过滤器,通过柱色谱法纯化受保护的(s)-2-(4-丙烯酰胺基苯基)-2-氨基乙酸,并蒸发残留的溶剂。将获得的(s)-2-(4-丙烯酰胺基苯基)-2-((叔丁氧羰基)氨基)乙酸(500mg,1.5mmol)溶于5ml二氯甲烷(dcm)中。添加三氟乙酸(tfa)(800μl,10.38mmol),并将该溶液在室温搅拌1小时。之后,减压去除溶剂,添加5ml dcm,并且再次减压去除溶剂。将该工序重复几次。最后,将产物溶于3ml dcm中,并用甲基叔丁基醚(mtbe)沉淀。将固体收集到玻璃过滤器上,并真空干燥,以获得产率为15%的纯丙烯酰基-4-氨基-l-苯丙氨酸。通过nmr验证化合物的结构。
[0193]
实施例9:合成(2s)-4-(丙烯酰氧基)吡咯烷-2-羧酸和(r)-3-(丙烯酰硫)-2-氨基丙酸
[0194]
如实施例4所述执行这些化合物的合成。对于(2s)-4-(丙烯酰氧基)吡咯烷-2-羧酸和(r)-3-(丙烯酰硫)-2-氨基丙酸,起始材料分别是4-羟基-l-脯氨酸和l-半胱氨酸。
[0195]
实施例10基于实施例8合成ahox-叠氮化物
[0196][0197]
将丙烯酰基-4-氨基-l-苯丙氨酸(4.0当量)的四氢呋喃溶液滴加到6-丙烯酰氨基-2-氨基己酸(1.0当量)和碳酸氢钠(2.0当量)在h2o的冷却溶液中。将溶液于0℃搅拌1小时,然后在室温下搅拌过夜。将thf从溶液中减压去除。然后用6m hcl将溶液酸化至ph为1,并用乙酸乙酯萃取(3次)。将合并的有机相经na2so4干燥,并减压浓缩。将所得油状膜溶于少量etoac中,并滴加到庚烷中。滤出细白色沉淀并在真空下干燥,从而得到所需的产物。
[0198]
实施例11:boc-g
(3)-raft试剂的合成
[0199]
步骤1:步骤1:合成raft-nhs中间体:
[0200][0201]
在0℃向如在tucker等人(acs macro letters(2017)6(4):452-457)中所述合成的乙基-raft(22.85g,102mmol,1.0当量)和1-羟基吡咯烷-2,5-二酮(12.89g,112mmol,1.1当量)在ch2cl2中的溶液中加入edc
·
hcl(21.48g,112mmol,1.1当量)。将反应混合物在室温下搅拌16小时。然后在n2流下将反应混合物部分蒸发(至总体积的约一半),并用acoet和双蒸水(ddh2o)稀释。将两相溶液转移到分液漏斗中,并且在萃取后,将有机相依次用ddh2o、nahco3的饱和水溶液(3x)、ddh2o(2x)和盐水洗涤。干燥有机相(na2so4),并减压去除所有挥发物。将残余物与正己烷一起研磨,并将所得黄色悬浮液过滤。将滤饼用正己烷洗涤。将黄
色固体减压干燥,并且将所得中间体(raft-nhs)不经进一步纯化即使用(31.8g,99.0mmol,97%)。所有分析数据均与文献值一致。yang等人,(2012)macromolecular rapid communications 33(22):1921-6。
[0202]
步骤2:合成raft-eda-boc中间体:
[0203][0204]
在-10℃向raft-nhs起始材料(1.22g,3.61mmol,1.0当量)在ch2cl2中的溶液中滴加叔丁基-(2-氨乙基)氨基甲酸酯(0.81g,5.0mmol,1.4当量)和et3n(1.0ml,7.2mmol,2.0当量)在ch2cl2中的溶液。将反应混合物在室温搅拌12小时。依次用nh4cl饱和水溶液(2
×
)、nahco3饱和水溶液(2
×
)和盐水洗涤有机混合物。干燥有机相(na2so4),并减压去除所有挥发物。将残余物从正庚烷和et2o的混合物中重结晶。将黄色晶体滤出,用正庚烷洗涤,并减压干燥,以得到下一中间体(raft-eda-boc,1.26g,3.44mmol,95%)。通过ms波谱法和nmr波谱法验证所获得的化合物的结构。
[0205]
步骤3:合成raft-eda-otf中间体:
[0206][0207]
将raft-eda-boc(1.25g,3.41mmol,1.0当量)在tfa中的冷溶液搅拌60分钟。然后将反应混合物用meoh和ch2cl2(1/2)稀释,并在n2流下部分地(占总体积的2/3)去除挥发物。所得raft-eda-otf被分离为黄色油状物(2.00g,3.29mmol,96%),其无需进一步纯化即用于下一步骤。通过ms波谱法和nmr波谱法验证所获得的化合物的结构。
[0208]
步骤4:boc-g
(3)-raft中间体的合成:
[0209][0210]
将boc-g
(3)
(bachem ag,bubendorf,switzerland)(697mg,2.41mmol,1.0当量)、1-羟基苯并三唑水合物(hobt水合物)(92.0mg,600μmol,0.25当量)和edc hcl(485mg,2.53mmol,1.05当量)在ch2cl2中的溶液在惰性气氛(n2)下在0℃搅拌30分钟。向该溶液依次滴加raft-eda-otf(917mg,2.41mmol,1.0当量)在ch2cl2中的溶液和dipea(2.13ml,12.5mmol,5.2当量)。将反应混合物在0℃下搅拌1小时,然后在室温下搅拌过夜。将反应混合物用ch2cl2稀释,并将有机混合物依次用nh4cl饱和溶液(3x)、nahco3饱和溶液、ddh2o和盐水洗涤。收集有机相,干燥(na2so4),并在减压下部分地去除挥发物(占总体积的2/3)。向所得溶液中添加etoac。然后将所得浑浊溶液在冰箱中存储过夜,得到黄色悬浮液,将该黄色悬浮液过滤,并用冷etoac洗涤滤饼。将该黄色固体减压干燥,以获得boc-g
(3)-raft试剂(396mg,736μmol,31%)。通过ms波谱法和nmr波谱法验证所获得的化合物的结构。
[0211]
在此给出的实施例被认为是用于合成raft试剂的一般工序,所述raft试剂是用ology-甘氨酸间隔基官能化的。更长或更短的间隔基团可以通过寡聚甘氨酸结构单元的交
换来合成。
[0212]
实施例12:raft-peg
(5)-nh3cl的合成
[0213][0214]
在0℃下,向raft-nhs起始材料(1.4mmol)的ch2cl2溶液中滴加boc-peg
(5)-ch2ch
2-nh2(1.4mmol)和et3n(1.5mmol)的ch2cl2溶液。然后将反应混合物在室温下搅拌过夜。将混合物蒸发并通过rp-18柱色谱(1:1acn:h2o)纯化,以得到raft-peg
(5)-boc(1.2mmol,88%)。根据质谱和nmr光谱数据确定结构归属。
[0215]
将raft-peg
(5)-boc(0.25mmol)在3m hcl中的冷溶液在etoac中搅拌120min。然后蒸发反应混合物,并通过rp-18柱色谱法(1:1acn:h2o 0.1%乙酸)纯化。分离所得的raft-peg
(5)-nh3cl作为黄色油状物(0.21mmol,84%)。根据质谱和nmr光谱数据确定结构归属。
[0216]
实施例13:使用nh
2-peg
(5)
raft合成nh
2-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)共聚物
[0217][0218]
向dma(100μl,970μmol,30当量)和ak-叠氮化物(69.7mg,259μmol,8当量)在0.1m nahco3中的溶液中连续加入nh
2-peg
(5)
raft(16.92mg,32.2μmol,1.0当量)和va044(3.14mg,9.7μmol,0.3当量)。将反应混合物在60℃下搅拌4小时。将反应混合物用ddh2o和二噁烷稀释。向该溶液中依次添加次膦酸(50w%,27μl,158μmol,5当量)、tea(22μl,158μmol,5当量)和aibn(1.6mg,9.5μmol,0.3当量)。将反应混合物在75℃下搅拌8小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得呈白色粉末的nh
2-peg
(5)-(dma
(45)-ak-叠氮化物
(4)
)(140mg,120μmol,两个步骤中85%)。使用以下方案,通过nmr波谱法和gpc调查所获得的化合物的结构:在洗脱缓冲液(含0.05%(w/v)nan3的去离子水)中制备含有3.33mg/ml共聚物的储备溶液,并滤过0.45μm注射器过滤器。随后,将0.4ml储备溶液注入gpc装置(1260infinity lc系统,agilent,santa clara,ca)的端口中。在洗脱缓冲液中以0.5ml/min的恒定流速执行色谱分析。将共聚物样品在suprema三柱系统(预柱,柱,粒径5μm;pss,mainz,germany)上分离,该suprema三柱系统放置于55℃的外部柱温箱中。通过ri(折射率)和紫外检测器分析共聚物。使用从pss(mainz,germany)获得的支链淀粉标准品建立校准曲线(10点),所述支链淀粉标准品包括以下10种聚合物(给出了mw、mn和pdi):(1)mw:342/mn:342,pdi 1.0;(2)mw:1320/mn:1080,pdi 1.23;(3)mw:6200/mn:5900,pdi 1.05;(4)mw:10000/mn:9200,pdi 1.09;(5)mw:21700/mn:20000,pdi 1.09;(6)mw:48800/mn 45500,pdi 1.07;(7)mw:113000/mn:100000,pdi 1.13;(8)mw:
210000/mn 189000,pdi 1.11;(9)mw:366000/mn 318000,pdi 1.15;(10)mw:805000/mn:636000,pdi:1.27。参照该标准估算经表征的共聚物的分子量。为此,基于通过软件pss wingpc unichrom v:8.1build 2827(pss;https://www.pss-polymer.com/)进行的gpc测量来确定聚合物的重均分子量(mw)、聚合物的数均分子量(mn)及其pdi。
[0219]
实施例14:nh
2-peg
(5)-(dma
(45)
ak-dota
(4)
)的合成
[0220][0221]
将nh
2-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)(20mg,3.45μmol)和螯合剂bcn-dota(chematech dijon,france)(24mg,34μmol)在0.1m nahco3中的溶液于35℃搅拌24小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得nh
2-peg
(5)-(dma
(45)
ak-dota
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0222]
实施例15:dbco-nh-peg
(5)-(dma
(45)
ak-dota
(4)
)的合成
[0223][0224]
将根据实施例14合成的nh
2-peg
(5)-(dma
(45)
ak-dota
(4)
)(10mg,1.15μmol)、dbco-nhs(3.96mg,9.2μmol)和tea(1.275μl,9.2μmol)在dmf中的溶液于25℃搅拌7小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得dbco-nh-peg
(5)-(dma
(45)
ak-dota
(4)
)。通过nmr光谱学研究所获得的化合物的结构。为了验证这种dbco基团的活性,将所得聚合物溶于dmf中,并在室温下加入过量的fam-叠氮化物,并将混合物搅拌4h。使用实施例13中给出的方案,用gpc验证所得化合物。
[0225]
实施例16:用于诊断和治疗性靶向过表达her2受体的癌细胞的经放射性标记的曲妥珠单抗-[nh-peg
(4)-三唑peg
(5)-(dma
(45)
ak-dota
(4)
)]2缀合物的合成
[0226][0227]
在肿瘤诊断中,原发性肿瘤或其转移的检出限对于患者的存活率至关重要,因为晚期肿瘤通常与不良预后相关联。使用放射性标记的肿瘤组织特异性抗体进行癌细胞的检测和后续疗法是放射医学的潜在有前途的方法。然而,由于事实上只有很少的放射性同位素可以附着至靶向部分/抗体上,并且感兴趣的放射性同位素具有短半衰期(通常比抗体的半衰期短),因此此类方法因低信噪比而受阻。因此,非常需要增加放射性同位素的货物载量。在该实施例中,描述了用于改善的肿瘤细胞检测和疗法的放射性标记的抗体-共聚物缀合物。
[0228]
通过实施例15中所述的工序合成的dbco官能化共聚物dbco-nh-peg
(5)-(dma
(45)-ak-dota
(4)
缀合至igg型癌细胞特异性抗体(用于靶向her2 癌细胞的曲妥珠单抗),该igg型癌细胞特异性抗体已经通过由dennler等人(bioconjugate chem.(2014)25:569-578)描述的方法在295位(q 295)中的谷氨酰胺处被叠氮基官能化。
[0229]
简而言之,该抗体被png酶f(merck kgaa,darmstadt,germany)去糖基化。将含有在pbs(ph 7.4)中每10μg曲妥珠单抗(carbosynth ltd,berkshir,uk)1单位酶的反应混合物在37℃下孵育过夜以激活q295。随后,将pbs(ph 8)中的去糖基化的曲妥珠单抗(6.6μm)与nh
2-peg
(4)-叠氮化物(click chemistry tools,scottsdale,usa)(80摩尔当量)和微生物转谷氨酰胺酶(mtg酶)(6u/ml,zedira,darmstadt,germany)一起于37℃孵育16h。在孵育后,通过添加mtg酶反应终止剂(zedira,darmstadt,germany)来阻断mtg酶的活性。为了去除过量的nh
2-peg
4-叠氮化物、mtg酶和残留的png酶f,通过使用ultra 4ml柱(100kda mwco,merck kgaa,darmstadt,germany)将反应混合物进行缓冲液交换(三次)到nh4oac(0.5m,ph 5.5)中。
[0230]
随后,通过以下方式来执行实际的点击反应:将曲妥珠单抗-(nh-peg
(4)-叠氮化物)2与3倍摩尔过量的dbco官能化聚合物于37℃孵育过夜,从而产生曲妥珠单抗-[nh-peg
(4)-三唑-peg
(5)-(dma
(45)
ak-dota
(4)
)]2。使用未修饰的曲妥珠单抗作为对照,通过sds page研究反应的成功。通过添加5μl 4x sds-page上样缓冲液 10%w/vβ-巯基乙醇(biorad,germany)并孵育(60min,,37℃,以600rpm恒定振荡)来终止反应混合物(20μl)。随后将样品在4-20%sds-page凝胶(tgx
tm
预制凝胶,biorad,germany)上于150v电泳40分钟,然后对凝胶进行考马斯亮蓝染色。这些实验揭示了抗体重链与共聚物的定量官能化。
[0231]
过量的聚合物和残留的未官能化的曲妥珠单抗可以通过尺寸排阻色谱法(size exclusion chromatography,sec)去除,并且可以合并含有所需产物的级分。
[0232]
于37℃用111-incl3(每μg曲妥珠单抗-[nh-peg
(4)-三唑-peg
(5)-(dma
(45)
ak-dota
(4)
)]
2 4mbq)执行对抗体-共聚物缀合物的放射性标记达1小时,在此之后通过在
superdex 75 10/300gl柱(ge healthcare,chicago,usa)上以0.5ml/min的流速运行的sec对铟-111标记的抗体-聚合物-缀合物进行纯化。合并主要峰级分。所得曲妥珠单抗-[nh-peg
(4)-三唑-peg
(5)-(dma
(45)
ak-dota-in-111
(4)
)]2可用于例如在乳腺癌、结肠癌或肺癌患者中通过正电子发射断层扫描(pet)检测her2 癌细胞,其灵敏度高于通过常规抗体-放射性同位素复合物所能达到的灵敏度。增加的灵敏度是由于与常规放射性标记的抗体相比,抗体-载体复合物所承载的-111货物增加。
[0233]
可以使用相同工序来制备负载有合适的放射性同位素(如镥-177)的治疗性抗体-共聚物缀合物[用177-lucl3代替上述工序中使用的111-incl3]。
[0234]
实施例17:使用nh
2-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)合成四嗪-nh-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)共聚物
[0235][0236]
将根据实施例13合成的nh
2-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)(3.57μmol)、四嗪-nhs(18μmol)和tea(3.57μmol)的dmf溶液于25℃搅拌8小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得四嗪-nh-peg
(5)-(dma
(45)
ak-叠氮化物
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0237]
实施例18:boc-g
(3)-raft预聚物的合成
[0238][0239]
向dma(1000μl,9704μmol,15当量)的二噁烷溶液中连续加入boc-g
3-raft(348mg,647μmol,1.0当量)和aibn(31.9mg,194μmol,0.3当量)。将反应混合物于70℃搅拌4h。将反应混合物用正己烷稀释,并且在正己烷洗涤后获得了呈黄色粉末的预聚物。使用以下方案,通过nmr波谱法和gpc调查所获得的化合物的结构:在洗脱缓冲液(含0.05%(w/v)nan3的去离子水)中制备含有3.33mg/ml共聚物的储备溶液,并滤过0.45μm注射器过滤器。随后,将0.4ml储备溶液注入gpc装置(1260infinity lc系统,agilent,santa clara,ca)的端口中。在洗脱缓冲液中以0.5ml/min的恒定流速执行色谱分析。将共聚物样品在suprema三柱系统(预柱,粒径5μm;pss,mainz,germany)上分离,该suprema三柱系统放置于55℃的外部柱温箱中。通过ri(折射率)和紫外检测器分析共聚物。使用支链淀粉标准品建立校准曲线(10点)。参照该标准品来估算表征的共聚物的分子量。
[0240]
实施例19:boc-g
(3)-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)的合成
[0241][0242]
向dma(116μl,1120μmol,30当量)、ak(30mg,150μmol,4当量)、ak-叠氮化物(42mg,150μmol,4当量)在ddh2o中的溶液中连续加入boc-g
(3)-raft预聚物(79mg,37μmol,1.0当量)和va044(3.6mg,11.2μmol,0.3当量)。将反应混合物在60℃下搅拌4小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得boc-g
(3)-(dma
(45)
ak
(4)
ak-叠氮化物(4))。使用实施例13中给出的方案通过nmr光谱法和gpc研究所得化合物的结构。
[0243]
实施例20:boc-g
(3)-(dma
(45)
ak
(4)
ak-dota
(4)
)的合成
[0244][0245]
向boc-g
3-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)(260mg,37μmol,1当量)的dmf溶液中加入dbco-dota(199mg,300μmol,8当量)的dmf溶液。将反应混合物在室温下搅拌4h,并用二噁烷稀释。向该溶液中依次加入次膦酸(50w%,32μl,185μmol,5当量)、tea(26μl,158μmol,5当量)和aibn(1.9mg,11.1μmol,0.3当量)。将反应混合物在75℃下搅拌8小时。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得呈白色粉末的boc-g
3-(dma
(45)
ak
(4)
ak-dota
(4)
)(198mg,22μmol,三个步骤中56%)。使用实施例13中给出的方案通过nmr光谱法和gpc研究所得化合物的结构。
[0246]
实施例21:boc-g
3-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)的合成
[0247][0248]
向boc-g
3-(dma
(45)
ak
(4)
ak-dota
(4)
)(14mg,1.5μmol,1当量)在ddh2o中的溶液中加入mmae-nhs(5当量)(细胞毒性剂)的dmso溶液,并于35℃搅拌24h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得boc-g
(3)-(dma
(45)
ak-mmae
(4)-ak-dota
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0249]
实施例22:dbco-g
3-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)的合成
[0250][0251]
将boc-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)(28mg,2μmol)溶解在tfa的dcm(1:1)溶液中。4h后,减压去除有机溶剂,以获得nh3cl-g
3-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)。向nh3cl-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)(14,1μmol,1当量)的dmf溶液中连续加入dbco-nhs(3.4mg,8μmol,8.0当量)和tea(1.1μl,8μmol,8.0当量)。将反应混合物于25℃搅拌4h。将所得混合物用ddh2o稀释,然后相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得dbco-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
。通过nmr光谱学研究所获得的化合物的结构。为了验证这种dbco基团的活性,将所得聚合物溶于dmf中,并加入fam-叠氮化物。使用实施例13中给出的方案,用gpc验证所得化合物。
[0252]
实施例23:用于诊断和治疗性靶向过表达her2受体的癌细胞的经放射性标记的曲妥珠单抗-[nh-peg
(4)-三唑-peg-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
]2缀合物的合成
[0253][0254]
大多数肿瘤疗法通过肿瘤大小的减小(ct、mrt扫描)进行定性监测,并且通常不执
行细胞毒性剂分布的详细表征。在一些情况下,将有用的是用非侵入性方法获得关于实际组织分布的信息。这可以通过将用于治疗方法的细胞毒素和用于监测的诊断放射性同位素结合到同一药物载体来实现。由于这种方法,抗体-聚合物缀合物的命运可以被可视化,并给出了关于潜在副作用的有用信息,例如在肝脏中的非预期代谢或在其他组织中的聚集。这种方法将尤其有利于对剂量发现和副作用进行表征的i期临床研究。
[0255]
通过实施例22中所述的工序合成的dbco官能化的共聚物(dbco-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
))缀合至igg型癌细胞特异性抗体(用于靶向her2 癌细胞的曲妥珠单抗),该igg型癌细胞特异性抗体通过由dennler等人(bioconjugate chem.(2014)25:569-578)描述的方法在295位(q 295)中的谷氨酰胺处被叠氮基官能化。
[0256]
简而言之,该抗体被png酶f(merck kgaa,darmstadt,germany)去糖基化。将含有在pbs(ph 7.4)中每10μg曲妥珠单抗(carbosynth ltd,berkshir,uk)1单位酶的反应混合物在37℃下孵育过夜以激活q295。随后,将pbs(ph 8)中的去糖基化的曲妥珠单抗(6.6μm)与nh
2-peg
4-叠氮化物(click chemistry tools,scottsdale,usa)(80摩尔当量)和微生物转谷氨酰胺酶(mtg酶)(6u/ml,zedira,darmstadt,germany)一起于37℃孵育16h。在孵育后,通过添加mtg酶反应终止剂(zedira,darmstadt,germany)来阻断mtg酶的活性。为了去除过量的nh
2-peg
4-叠氮化物、mtg酶和残留的png酶f,通过使用ultra 4ml柱(100kda mwco,merck kgaa,darmstadt,germany)将反应混合物进行缓冲液交换(三次)到nh4oac(0.5m,ph 5.5)中。
[0257]
随后,通过以下方式来执行实际的点击反应:将曲妥珠单抗-(nh-peg
4-叠氮化物)2与3倍摩尔过量的dbco官能化聚合物于37℃孵育过夜,从而产生曲妥珠单抗-[dbco-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota(4)]2。使用未修饰的曲妥珠单抗作为对照,通过sds page研究反应的成功。通过添加5μl 4x sds-page上样缓冲液 10%w/vβ-巯基乙醇(biorad,germany)并孵育(60min,37℃,以600rpm恒定振荡)来终止反应混合物(20μl)。随后将样品在4-20%sds-page凝胶(tgx
tm
预制凝胶,biorad,germany)上于150v电泳40分钟,然后对凝胶进行考马斯亮蓝染色。
[0258]
过量的聚合物和残余的非官能化的曲妥珠单抗可以通过尺寸排阻色谱法(sec)并合并含有完全官能化抗体的级分来去除。
[0259]
于37℃用111-incl3(每μg曲妥珠单抗-[dbco-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-dota(4)]
2 4mbq)执行对抗体-共聚物缀合物的放射性标记达1小时,在此之后通过在superdex 75 10/300 gl柱(ge healthcare,chicago,usa)上以0.5ml/min的流速运行的sec对铟-111标记的抗体-聚合物-缀合物进行纯化。合并主要峰级分。所得曲妥珠单抗-[dbco-nh-g
(3)-(dma
(45)
ak-mmae
(4)
ak-叠氮化物-(dota-in-111)4]2可用于例如在乳腺癌、结肠癌或肺癌患者中通过正电子发射断层扫描(pet)检测her2 癌细胞,其灵敏度高于通过常规抗体-放射性同位素复合物所能达到的灵敏度。增加的灵敏度是由于与常规放射性标记的抗体相比,抗体-载体复合物所承载的-111货物增加。
[0260]
实施例24:四嗪-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)
[0261][0262]
向boc-g
(3)-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)(14mg,1.5μmol,1当量)在ddh2o中的溶液中加入dota-nhs(6μmol,4当量)的dmso溶液,并于35℃搅拌24h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得boc-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)。随后将boc-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)(2μmol)溶解在tfa的dcm(1:1)溶液中。4小时后,减压去除有机溶剂,以获得nh
2-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)。向nh
2-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)(14,1μmol)的dmf溶液中连续加入四嗪-nhs(5μmol)和tea(2μmol)。将反应混合物于25℃搅拌8h。将所得混合物用ddh2o稀释,然后相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得四嗪-nh-g
(3)-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0263]
实施例25:hs-(dma
(45)
ak-叠氮化物
(4)
)的合成
[0264][0265]
向dma(116μl,1120μmol,45当量)、ak-叠氮化物(42mg,150μmol,4当量)的ddh2o溶液中连续加入乙基-raft(参见实施例11)(5.6mg,24.9μmol,1.0当量)和va044(3.6mg,11.2μmol,0.3当量)。反应混合物于60℃搅拌4h。将反应混合物在室温下搅拌4h。将环己胺(493μl,4977μmol,200当量)加入到反应混合物中,并于30℃搅拌3h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得呈白色粉末的hs-(dma
(45)
ak-叠氮化物
(4)
)(120mg,20μmol,三个步骤中81%)。使用实施例13的方案通过nmr光谱法和gpc研究所得化合物的结构。
[0266]
实施例26:hs-(dma
(45)
ak-dota
(4)
)的合成
[0267][0268]
向hs-(dma
(45
ak-叠氮化物
(4)
)(20mg,3.5μmol,1当量)在ddh2o中的溶液中加入dbco-dota(19.0mg,24μmol,8当量)的dmso溶液,并于35℃搅拌24h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得hs-(dma
(45)
ak-dota
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0269]
实施例27 dbco-(dma
(45)
ak-dota
(4)
)的合成
[0270][0271]
向hs-(dma
(45)
ak-dota
(4)
)(28mg,3μmol)的dmf溶液中加入mc-dbco(11.61mg,14.4μmol,8.0当量)。在4h后,将反应混合物用ddh2o稀释,然后相对于0.1m nh4hco3透析(mwco 3.5kda),并将保留物冷冻干燥以获得dbco-(dma
(45)
ak-dota
(4)
。通过nmr光谱学研究所获得的化合物的结构。为了验证这种dbco基团的活性,将所得聚合物溶于dmf中,并加入fam-叠氮化物。使用以下方案,用gpc验证所得化合物:在洗脱缓冲液(含0.05%(w/v)nan3的去离子水)中制备含有3.33mg/ml共聚物的储备溶液,并滤过0.45μm注射器过滤器。随后,将0.4ml储备溶液注入gpc装置(1260infinity lc系统,agilent,santa clara,ca)的端口中。在洗脱缓冲液中以0.5ml/min的恒定流速执行色谱分析。将共聚物样品在suprema三柱系统(预柱,粒径5μm;pss,mainz,germany)上分离,该suprema三柱系统放置于
55℃的外部柱温箱中。通过ri(折射率)和紫外检测器分析共聚物。使用支链淀粉标准品建立校准曲线(10点)。参照该标准估算经表征的共聚物的分子量。在该测试中,495nm的信号(fam)和共聚物的ri信号在重叠处显示匹配,表明该共聚物被活性dbco头基官能化。
[0272]
实施例28:hs-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)的合成
[0273][0274]
向dma(116μl,1120μmol,45当量)、ak(30mg,150μmol,4当量)、ak-叠氮化物(42mg,150μmol,4当量)的ddh2o溶液中连续加入乙基-raft(参见实施例11)(5.6mg,24.9μmol,1.0当量)和va044(3.6mg,11.2μmol,0.3当量)。反应混合物于60℃搅拌4h。将反应混合物在室温下搅拌4h。将环己胺(493μl,4977μmol,200当量)加入到反应混合物中,并于30℃搅拌3h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得呈白色粉末的hs-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)(150mg,23μmol,三个步骤中88%)。使用实施例13的方案通过nmr光谱法和gpc验证所得化合物的结构。
[0275]
实施例29:hs-(dma
(45)
ak
(4)
ak-dota
(4)
)的合成
[0276][0277]
向hs-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)(20mg,3μmol,1当量)在ddh2o中的溶液中加入dbco-dota(16.3mg,24μmol,8当量)的dmso溶液,并于35℃搅拌24h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得hs-(dma
(45)
ak
(4)
ak-dota
(4)
)。通过nmr光谱学研究所获得的化合物的结构。
[0278]
实施例30:将hs-(dma
(45)
ak
(4)
ak-dota
(4)
)与mmae偶联
[0279][0280]
向hs-(dma
(45)
ak
(4)
ak-dota
(4)
)(14mg,1.5μmol,1当量)在ddh2o中的溶液中加入mmae-nhs(15.16mg,12μmol,8当量)的dmso溶液,并于35℃搅拌24h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得hs-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)。可以通过nmr光谱学研究所获得的化合物的结构。
[0281]
实施例31:dbco-dma
(45)
ak-mmae
(4)
ak-dota
(4)
)的合成
[0282][0283]
向hs-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
)(28mg,1.8μmol)的dmf溶液中连续加入mc-dbco(6.1mg,14.4μmol,8.0当量)。在4h后,将反应混合物用ddh2o稀释,然后相对于0.1m nh4hco3透析(mwco 3.5kda),并将保留物冷冻干燥以获得dbco-mc-s-(dma
(45)
ak-mmae
(4)
ak-dota
(4)
。通过nmr光谱学研究所获得的化合物的结构。为了验证dbco-基团的活性,将少量的共聚物样品溶于dmf中,加入fam-叠氮化物,并将混合物于37℃孵育4h。之后,可以使用实施例13的方案通过gpc分析如此官能化的共聚物,并检测ri信号和uv(495nm)。在该测试中,495nm的信号(fam)和共聚物的ri信号在重叠处显示匹配,表明该共聚物被活性dbco头基官能化。
[0284]
实施例32:四嗪-(dma
(45)
ak-叠氮化物
(4)
)的合成
[0285][0286]
向hs-(dma
(45)
ak-叠氮化物
(4)
)(1当量)的dmf溶液中加入四嗪-peg
(4)-mc(8.0当量)。在4h后,将反应混合物用ddh2o稀释,然后相对于0.1m nh4hco3透析(mwco 3.5kda),并将保留物冷冻干燥以获得四嗪-(dma
(45)
ak-叠氮化物
(4)
)。
[0287]
实施例33:四嗪-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)的合成
[0288][0289]
向hs-(dma
(45)
ak
(4)
ak-叠氮化物
(4)
)(1当量)在ddh2o中的溶液中加入dota-nhs(6μmol,4当量)的dmso溶液,然后将混合物于35℃搅拌24h。然后将混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得hs-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)。
[0290]
向hs-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)(1当量)的dmf溶液中加入四嗪-peg
(4)-mc(8.0当量)。在4h后,将反应混合物用ddh2o稀释,然后相对于0.1m nh4hco3透析(mwco 3.5kda),并将保留物冷冻干燥以获得四嗪-(dma
(45)
ak-dota
(4)
ak-叠氮化物
(4)
)。
[0291]
实施例34:用于诊断和治疗性靶向过表达her2受体的癌细胞的经放射性标记的曲妥珠单抗-叠氮化物-共聚物(实施例17、实施例24、实施例32和实施例33)缀合物的合成
[0292]
通过实施例17、实施例24、实施例32和实施例33中所述的工序中的一个工序合成的四嗪官能化共聚物缀合至igg型癌细胞特异性抗体(例如,用于靶向her2 癌细胞的曲妥珠单抗),该igg型癌细胞特异性抗体已使用tco-peg
(3)-胺作为底物,通过由dennler等人(bioconjugate chem.(2014)25:569-578)描述的方法在295位(q 295)中的谷氨酰胺处被tco基团官能化。
[0293]
简而言之,该抗体被png酶f(merck kgaa,darmstadt,germany)去糖基化。将含有在pbs(ph 7.4)中每10μg曲妥珠单抗(carbosynth ltd,berkshir,uk)1单位酶的反应混合物在37℃下孵育过夜以激活q295。随后,将pbs(ph 8)中的去糖基化的曲妥珠单抗(6.6μm)与tco-peg
(3)-胺(80摩尔当量)和微生物转谷氨酰胺酶(mtg酶)(6u/ml,zedira,darmstadt,germany)一起于37℃孵育16h。在孵育后,通过添加mtg酶反应终止剂(zedira,darmstadt,germany)来阻断mtg酶的活性。为了去除过量的tco-peg
(3)-胺、mtg酶和残留的png酶f,通过使用ultra 4ml柱(100kda mwco,merck kgaa,darmstadt,germany)将反应混合物进行缓冲液交换(三次)到nh4oac(0.5m,ph 5.5)中。
[0294]
随后,通过以下方式来执行实际的点击反应:将曲妥珠单抗-(nh-peg
(3)-tco)
(2)
与3倍摩尔过量的四嗪官能化聚合物(实施例17、实施例24、实施例32或实施例33)于37℃过夜孵育,从而产生与实施例17、实施例24、实施例32和实施例33中合成的共聚物偶联的曲妥珠单抗。使用未修饰的曲妥珠单抗作为对照,通过sds-page研究反应的成功。为此,通过加入5μl 4x sds-page上样缓冲液 10%w/vβ-巯基乙醇(biorad,germany)并于37℃孵育60min(以600rpm持续振荡)来停止反应样本(20μl)。随后将样品在4-20%sds-page凝胶(tgx
tm
预制凝胶,biorad,germany)上于150v电泳40分钟,然后对凝胶进行考马斯亮蓝染色。
[0295]
实施例35:用细胞毒性药物装载曲妥珠单抗-叠氮化物-共聚物缀合物
[0296]
向根据实施例34合成的曲妥珠单抗-叠氮化物-共聚物的pbs溶液中加入dbco-peg
(3)-vc-pab-mmae(lucerna-chem,lucerne,3当量/ak-叠氮化物单体),并将混合物于37℃孵育12小时。使用未装载的曲妥珠单抗-(叠氮化物-共聚物)2作为对照,根据以上实施例中给出的工序,通过sds-page和gpc研究反应的成功。
[0297]
实施例36:dbco-cf3(模型有效负载)的合成
[0298][0299]
于0℃向市售dbco-co2h(1353016-70-2,100mg,0.328mmol)在干dcm(6ml)中的溶液中加入dipea(0.200ml,1.146mmol)和pybop(170mg,0.328mmol)。将混合物于0℃搅拌30min,并将2,2,2-三氟乙胺(0.039ml,0.491mmol)分批加入混合物中。然后将反应混合物于0℃搅拌2h,并用khso4水溶液稀释。然后将水相用dcm(3
×
20ml)萃取,并将合并的有机相用盐水洗涤并经na2so4干燥。然后减压去除挥发物,并将残余物通过使用1.2hept/1etoac作为洗脱剂的硅胶色谱柱纯化。分离出了呈白色粉末的产物(125mg,0.324mmol,99%)。
[0300]
实施例37:通过聚合物类似反应合成hs-(dma
(55)-ak-(6-叠氮基己酰基)
(4)
)
[0301][0302]
向通过使用乙基-raft(1当量)作为转移试剂(200mg,31μmol)进行dma(55当量)和ak(4当量)的raft聚合(类似的详细方案参见实施例25)获得的hs-(dma
(55)-ak
(4)
)在dmf(5ml)中的溶液中加入1(6-叠氮己酰基)吡咯烷-2,5-二酮(75mg,314μmol)和tea(44μl,314μmol)。将反应混合物在室温下搅拌16h,用ddh2o(12ml)稀释,并相对于ddh2o(10l)、nh4hco3水溶液(0.1m,3l)和ddh2o(10l)透析。将保留物冻干(-78℃,0.010毫巴)。通过1h/
13
c-nmr光谱学研究获得的构建体的结构,并在标题化合物用dbco-cf3衍生化后,通过
19
f-nmr光谱估计dar(=4.2)。
[0303]
实施例38:经由聚合物类似反应合成模型化合物hs-(dma
(55)-(ak-三嗪-cf3)
(1)-(ak-乙酰基-cf3)
(3)
[0304][0305]
向hs-(dma
(55)-ak-叠氮化物
(4)
)在dmf-d6(0.7ml)中的溶液中加入1-(6-叠氮己酰基)吡咯烷-2,5-二酮(5.98mg,25μmol)、1-(3,3,3-三氟丙酰基)吡咯烷-2,5-二酮(5.25mg,25μmol)(两种物质同时加入)和三乙胺(10.51μl,75μmol)。将混合物在室温下搅拌4h,然后加入dbco-cf3(19.40mg,50μmol),并将反应混合物搅拌过夜。然后通过超滤纯化聚合物(2000mwco,重悬并用3
×
10ml ddh2o过滤)。然后将保留物冻干以获得白色粉末(35mg,4.84μmol,77%)。通过
19
f-nmr光谱分析白色残余物。
[0306]
实施例39:模型化合物甲基四嗪-peg4-琥珀酸-s
‑‑
(dma
(55)-(ak-三嗪-cf3)
(1)-(ak-乙酰基-cf3)
(3)
的合成
[0307][0308]
向hs-(dma
(55)-(ak-三嗪-cf3)
(1)-(ak-乙酰基-cf3)
(3)
在dmf-d6(0.7ml)中的溶液
中加入四嗪-peg
(4)-mc(8.0当量)。在4h后,将反应混合物用ddh2o稀释,然后相对于0.1m nh4hco3透析(mwco 3.5kda),并将保留物冷冻干燥以获得甲基四嗪-peg
4-琥珀酸-s-(dma
(55)-(ak-三嗪-cf3)
(1)-(ak-乙酰基-cf3)
(3)
。
[0309]
实施例40:hs-(hpa
(55)-ak
(4)
)的合成
[0310]
这种化合物是类似于共聚物hs-(dma
(55)-ak-叠氮化物
(4)
)(参见实施例37的第一部分),使用按照fairbanks及其同事的方案(dx.doi.org/10.1021/bm500654q)获得的hpa(2-羟丙基)丙烯酰胺)(55当量)作为dma单体的替代品获得的。
[0311]
实施例41:hs-(heaa
53)
ak
(4)
)的合成
[0312][0313]
向heaa(368μl,3540μmol,53当量)、ak(54mg,267μmol,4当量)的ddh2o溶液中连续加入乙基-raft(参见实施例11)(15mg,67μmol,1.0当量)和va044(6.5mg,20μmol,0.3当量)。反应混合物于60℃搅拌4h。将反应混合物在室温下搅拌4h。将环己胺(1533μl,13.37mmol,200当量)加入到反应混合物中,并于30℃搅拌3h。然后将所得混合物相对于ddh2o进行渗析(mwco 3.5kda),并将保留物冷冻干燥,以获得呈白色粉末的hs-(heaa
(53)
ak
(4)
)(395mg,55.6μmol,三个步骤中83%)。使用实施例13的方案通过nmr光谱法和gpc研究所得化合物的结构。
[0314]
在以下编号的项目中公开了本发明的其他方面和/或实施方案。
[0315]
1.一种含有第一有效负载分子的多个拷贝的共聚物,所述共聚物可通过以下步骤制得:
[0316]
(a)使反应混合物聚合,所述反应混合物包含
[0317]
(1)含有叠氮基部分的式i的共-主要单体,
[0318][0319]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-;l是在生理条件下可分裂或不可分裂的连接基团/间隔基团。
[0320]
(2)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸
部分或叠氮基部分,和
[0321]
(3)用于生成自由基的引发剂体系,所述聚合产生共聚物;以及
[0322]
(b)将所述第一有效负载分子偶联至在步骤(a)的所述共聚物中包含的所述叠氮基部分。
[0323]
2.根据项目1所述的共聚物,其中所述反应混合物进一步包含式ii的共-主要单体或式iii的共-主要单体的一者或两者,
[0324][0325]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;y为h或-co-c
nh2n 1
(其中n=1-8);z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-,
[0326][0327][0328]
其中:r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;z为h(如果a为o)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-。
[0329]
3.根据项目2所述的共聚物,其中在步骤(a)或(b)之后,另一步骤包括将含反应性基团的第二有效负载分子偶联至式ii或式iii的共-主要单体的一者或两者,其中在所述式ii的单体中的y和z中的一者或两者是h,或者在所述式iii的单体中z是h。
[0330]
4.根据项目1-3中任一项所述的共聚物,其中所述反应混合物进一步包含用于控制共聚的raft试剂。
[0331]
5.根据项目4所述的共聚物,其中步骤(a)分成两个连续的聚合反应,
[0332]
其中所述第一聚合反应在第一反应混合物中进行,所述第一反应混合物包含不含氨基酸部分或叠氮基部分的可聚合主要单体、用于控制所述共聚的raft试剂、和用于生成自由基的引发剂体系,所述聚合产生raft预聚物,并且
[0333]
其中所述第二聚合反应在第二反应混合物中进行,所述第二反应混合物包含所述第一聚合反应的所述raft预聚物、式i的共-主要单体和用于生成自由基的引发剂体系。
[0334]
6.根据项目5所述的共聚物,其中所述第二反应混合物进一步包含式ii和iii中的一者或两者的共-主要单体和不含氨基酸部分或叠氮基部分的可聚合主要单体。
[0335]
7.根据项目4-6中任一项所述的共聚物,其中所述raft试剂含有反应性基团,或者在步骤(a)之后被转化以提供反应性基团。
[0336]
8.根据项目4-7所述的共聚物,其中所述raft试剂含有2-30个单位的单分散间隔基团。
[0337]
9.根据项目7-8中任一项所述的共聚物,其中在步骤(b)之前,将细胞类型特异性或组织类型特异性靶向部分偶联至所述反应性基团。
[0338]
10.根据项目7-8中任一项所述的共聚物,其中在步骤(b)之后,将细胞类型特异性或组织类型特异性靶向部分偶联至所述反应性基团。
[0339]
11.根据项目1-10中任一项所述的共聚物,其中所述第一有效负载分子是螯合剂,所述共聚物暴露于活性剂,并且所述活性剂被所述螯合剂捕获。
[0340]
12.根据项目1-11中任一项所述的共聚物,其中所述共聚物的平均分子量为5,000道尔顿至80,000道尔顿。
[0341]
13.根据项目1-11中任一项所述的共聚物,其中所述共聚物的平均分子量为5,000道尔顿至40,000道尔顿。
[0342]
14.根据项目1-11中任一项所述的共聚物,其中所述共聚物的平均分子量为5,000道尔顿至20,000道尔顿。
[0343]
15.根据项目3所述的共聚物,其中所述反应混合物含有
[0344]
(1)式i的共-主要单体,
[0345]
(2)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸部分或叠氮基部分,
[0346]
(3)任选地,用于控制自由基聚合的试剂,以及
[0347]
(3)用于生成自由基的引发剂体系;并且
[0348]
所述共聚物含有2-12个分子的所述式i的共-主要单体。
[0349]
16.根据项目15所述的共聚物,其中所述共聚物含有2-8个分子的所述式i的共-主要单体。
[0350]
17.根据项目15所述的共聚物,其中所述共聚物含有2-6个分子的所述式i的共-主要单体。
[0351]
18.根据项目2所述的共聚物,其中所述反应混合物含有
[0352]
(1)式i的共-主要单体,
[0353]
(2)式ii或iii中的一者或两者的共-主要单体,
[0354]
(3)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸部分或叠氮基部分,
[0355]
(4)任选地,用于控制自由基聚合的试剂,以及
[0356]
(5)用于生成自由基的引发剂体系;并且
[0357]
所述共聚物含有10-50个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0358]
19.根据项目18所述的共聚物,其中所述共聚物含有10-40个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0359]
20.根据项目18所述的共聚物,其中所述共聚物含有10-30个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0360]
21.根据项目3所述的共聚物,其中所述反应混合物含有
[0361]
(1)式i的共-主要单体,
[0362]
(2)式ii或iii中的一者或两者的共-主要单体,
[0363]
(3)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸部分或叠氮基部分,
[0364]
(4)任选地,用于控制自由基聚合的试剂,以及
[0365]
(5)用于生成自由基的引发剂体系,所述聚合产生共聚物;并且
[0366]
所述共聚物含有4-20个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0367]
22.根据项目18所述的共聚物,其中所述共聚物含有4-15个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0368]
23.根据项目18所述的共聚物,其中所述共聚物含有4-10个分子的所述式i至iii的共-主要单体中的任一共-主要单体。
[0369]
24.一种药物组合物,所述药物组合物包含有效量的根据项目1-23中任一项所述的共聚物和药学上可接受的载体或赋形剂。
[0370]
25.根据项目24所述的药物组合物用于治疗受试者的癌症或另一种疾病或病症的用途,所述用途包括将所述药物组合物施用给所述受试者。
[0371]
在以下编号的段落中公开了本发明的其他方面和/或实施方案。
[0372]
1.一种含有第一有效负载分子的多个拷贝的共聚物,所述共聚物可通过以下步骤制得:
[0373]
(a)使反应混合物聚合,所述反应混合物包含
[0374]
(1)式i的共-主要单体,
[0375][0376]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;y为h;z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-,
[0377]
(2)可聚合主要单体,所述单体的特征在于具有至少一个乙烯基并且不含氨基酸部分或叠氮基部分,
[0378]
(3)任选的用于控制共聚的raft试剂,和
[0379]
(4)用于生成自由基的引发剂体系,所述聚合产生共聚物;并且
[0380]
(b)用含有连接基团和叠氮基部分的胺反应剂处理步骤(a)的所述共聚物,以及
[0381]
(c)将所述第一有效负载分子偶联至在步骤(b)的所述共聚物中包含的所述叠氮基部分。
[0382]
2.根据段落1所述的共聚物,其中所述反应混合物进一步包含式iii的共-主要单体,
[0383][0384]
其中:r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;z为h(如果a为o)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-。
[0385]
3.根据段落1或2所述的共聚物,其中l是-co-c
nh2n
(其中n=1-10)或-co-pegn(其中n=1-14),或者其中l是-co-缬氨酸-瓜氨酸-pabc或其变体、缬氨酸-赖氨酸、缬氨酸-丙氨酸、缬氨酸-精氨酸、谷氨酸-缬氨酸-瓜氨酸。
[0386]
4.根据段落1至3中任一项所述的共聚物,其中所述可聚合主要单体是n,n-二甲基丙烯酰胺、n-异丁基丙烯酰胺、n-叔丁基-丙烯酰胺、n-羟乙基-丙烯酰胺、n-(2-羟丙基)-丙烯酰胺、n-(3-羟丙基)-丙烯酰胺、n-(3-羟丙基)-甲基丙烯酰胺、n-(2-羟丙基)-甲基丙烯酰胺、n-(3-氨基丙基)-丙烯酰胺盐酸盐、或n-(3-氨基丙基)-甲基丙烯酰胺盐酸盐。
[0387]
或者其中所述可聚合主要单体是甲基丙烯酸、丙烯酸2-羟乙酯、丙烯酸2-羟丙酯、丙烯酸3-羟丙酯、丙烯酸2-羟基-1-甲基乙基酯、丙烯酸2-氨基乙酯盐酸盐、甲基丙烯酸3-羟丙酯、甲基丙烯酸2-羟基-1-甲基乙基酯、甲基丙烯酸2-羟乙酯、甲基丙烯酸2-羟丙酯或甲基丙烯酸2-氨基乙酯盐酸盐。
[0388]
5.一种包含具有下式的重复单元的共聚物
[0389][0390]
其中r为-h、-ch3、-ch
2-ch3或-(ch2)
2-ch3;x为-nh(ch2)
4-、-nh(ch2)
3-、-o-c6h
4-ch
2-、-o-ch
2-、-o-ch(ch3)-、-s-ch
2-或-nh-c6h
4-ch
2-;z为h(如果a为-o-)或-c
nh2n 1
(其中n=1-8);并且a为-o-或-nh-;l是在生理条件下可分裂或不可分裂的连接基团/间隔基团。
[0391]
6.一种包含具有下式的重复单元的共聚物:
8);并且a为-o-或-nh-。
[0400]
8.根据段落7所述的共聚物,其中y是h和/或z是h。
[0401]
9.根据段落7所述的共聚物,其中y和/或z包含第二有效负载分子。
[0402]
10.根据段落5至9中任一项所述的共聚物,其中l是-co-c
nh2n
(其中n=1-10)或-co-pegn(其中n=1-14),或者其中l是-co-缬氨酸-瓜氨酸-pabc或其变体、缬氨酸-赖氨酸、缬氨酸-丙氨酸、缬氨酸-精氨酸、谷氨酸-缬氨酸-瓜氨酸。
[0403]
11.根据段落5至10中任一项所述的共聚物,所述共聚物还包含通过n,n-二甲基丙烯酰胺、n-异丁基丙烯酰胺、n-叔丁基丙烯酰胺、n-羟乙基丙烯酰胺、n-(2-羟丙基)-丙烯酰胺、n-(3-羟丙基)-丙烯酰胺、n-(3-羟丙基)-甲基丙烯酰胺、n-(2-羟丙基)-甲基丙烯酰胺、n-(3-氨基丙基)-丙烯酰胺盐酸盐、或n-(3-氨基丙基)-甲基丙烯酰胺盐酸盐的聚合获得的重复单元,或者通过甲基丙烯酸、2-羟乙基丙烯酸酯、2-羟丙基丙烯酸酯、3-羟丙基丙烯酸酯、2-羟基-1-甲基乙基-丙烯酸酯、2-氨基乙基丙烯酸酯盐酸盐、3-羟丙基甲基丙烯酸酯、2-羟基-1-甲基乙基甲基丙烯酸酯、2-羟乙基甲基丙烯酸酯、2-羟丙基甲基丙烯酸酯或2-氨基乙基甲基丙烯酸酯盐酸盐的聚合获得的重复单元。
[0404]
12.根据段落6至11中任一项所述的共聚物,其中不存在式(r2)和(r3)的重复单元,根据式(r1)的重复单元的平均数为2至12,优选地2至8,更优选地1至6。
[0405]
13.根据段落5至11中任一项所述的共聚物,其中式(r2)或(r3)的重复单元未被官能化,并且其中根据式(r1)、(r2)或(r3)的重复单元的平均数为10至50,优选地10至40,更优选地10至30。
[0406]
14.根据段落5至11中任一项所述的共聚物,其中式(r2)或(r3)的重复单元用第二有效负载分子官能化,并且其中根据式(r1)、(r2)或(r3)的重复单元的平均数为4至20,优选地4至15,更优选地4至10。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。