1.本发明涉及一种制备佐匹克隆杂质的方法,特别是涉及一种制备吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的方法。
技术背景
[0002]
佐匹克隆(zopiclone)化学名称为6-(5-氯吡啶-2-基)-7-[(4-甲基哌嗪-1-基)羰氧基]-5,6-二氢吡咯[3.4-b]吡嗪-5-酮,为吡咯烷酮类化合物,是由法国罗纳普朗克乐安公司(rhono-poulene rorer)开发的第三代镇静催眠药,系抑制性神经递质γ-氨基丁酸(gaba)受体激动剂,具有催眠、镇静、肌肉松弛、抗焦虑和抗惊厥作用,主要用于各种原因导致失眠性的短期治疗。
[0003]
右佐匹克隆(eszopiclone)是由美国seprator公司开发的快速短效非苯二氮类镇静安眠药,于2005年4月在美国上市,属于佐匹克隆的(s)异构体。其结构与作用与苯二氮草卓类有明显区别。右佐匹克隆与苯二氮草卓类结合于相同的受体与部位,但作用区域不同,与苯二氮草卓类相比,右佐匹克隆的作用时间更加迅速,药效更强,成瘾性更低。
[0004]
对于药品中的工艺杂质,应在原料合成阶段重点关注产品中可能出现的各类潜在杂质;当最终合成路线确定后应重点分析杂质的去除途径,确定生产过程中的关键质控点;随着工艺过程的不断成熟,再开展未知杂质的结构确认工作,并开发新的分析方法确定是否有潜在杂质的存在。
[0005]
cn101058581a公开了一种右佐匹克隆中间体i的制备方法,以3-(5-氯吡啶-2-氨基)甲酰基吡嗪-2-羧酸ii为原料,经过以下路线制备而成:
[0006][0007]
其中以氯甲酸酯为羧基活化剂,反应先经过中间体混酐(其结构见式iii)进一步形成中间体i。
[0008]
本发明人发现,上述反应中,如果存在原料2-氨基-5-氯吡啶残留,该原料可能与混酐中间体iii反应生成杂质吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]。
[0009]
[0010]
目前,国内文献中未见该杂质的相关报导,对药品杂质谱的控制是保证药品安全有效的重要措施,也是提升国产药品质量的关键环节,为了便于佐匹克隆的工艺研究和质量控制提供高纯度的杂质对照品,迫切需要提供一种简便的、低成本的制备该杂质的方法。
[0011]
本文发明了一种制备佐匹克隆杂质的方法,特别是涉及一种制备吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的方法。
技术实现要素:
[0012]
本发明提供了一种制备佐匹克隆杂质的方法,特别是涉及一种制备吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的方法。通过该方法,合成出了佐匹克隆工艺开发和质量控制过程中出现的杂质吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺],通过本发明的方法得到在上述杂质,纯度高,可以用作杂质对照品。
[0013]
本发明涉及一种吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的合成方法,是通过以下技术方案实现的,包括以下步骤:
[0014][0015]
1)以3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸(化合物g)为原料,在有机溶剂中,加入三乙胺、edci、hobt、2-氨基-5-氯吡啶进行缩合反应,搅拌4-18小时;
[0016]
2)反应结束后,将反应液倒入水中,萃取,分离;
[0017]
3)分离物过纯化柱纯化后得到吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]纯品。
[0018]
其中,步骤1)中:edci为1-(3-二甲胺基丙基)-3-乙基碳二亚胺,是缩合剂;
[0019]
hobt为1-羟基苯并三唑,结构式如下,也是常用于酰胺缩合中的催化剂。
[0020][0021]
所述3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸、三乙胺,edci,hobt和2-氨基-5-氯吡啶的摩尔比为1:1.5-2.5:1.0-1.5:1.0-1.5:1,优选为1:2:1.2:1.2:1。
[0022]
所述有机溶剂是本领域常用的或者常规的有机溶剂,选自但不限于下述溶剂中的一种或者多种:二氯甲烷(沸点是39.75℃)、氯仿(沸点61.15℃)、n,n-二甲基甲酰胺(dmf,沸点152.8℃)或乙腈(沸点81.6℃)等,优选二氯甲烷。
[0023]
所述有机溶剂的用量是3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸重量的2-30倍体积,优选的是5-20倍,优选为6-10倍。(重量和体积单位相对应,如重量为g,体积为ml,重量为kg,体积为l)
[0024]
所述反应温度为20℃至溶剂体系的沸点,优选于20℃到50℃,或优选为20到100
℃,进一步优选20到42℃。
[0025]
所述反应时间为10-18小时。
[0026]
其中,步骤2)中:萃取选用有机溶剂和饱和食盐水依次进行。水相用有机溶剂萃取2-4次,有机相用饱和食盐水洗涤1-2次。其中有机溶剂为二氯甲烷。
[0027]
其中,步骤3)中:所用层析柱,条件为:填充剂:200-300目硅胶,洗脱剂:石油醚
‑‑‑
石油醚/乙酸乙酯=10/1(从纯石油醚开始,然后逐步添加乙酸乙酯,直到石油醚和乙酸乙酯的体积比为10:1)。
[0028]
经过以上步骤,可以得到收率85%以上,纯度为99%以上的杂质化合物吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]。
[0029]
本发明杂质吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺],其含量和纯度的检测方法。步骤如下:
[0030]
取本品,用流动相制成每1ml约含1mg的溶液,作为供试品溶液。照高效液相色谱法,用十八烷基硅烷键和硅胶为填充剂(waters symmetry shield c18柱,4.6*250mm,5μm,端基封尾或效能相当的色谱柱);以乙腈-缓冲盐(取8.1g十二烷基硫酸钠与1.6g磷酸二氢钠,加水溶解并稀释至1000ml)(538:1000),用10%磷酸溶液调ph至4.0为流动相,检测波长为303nm;流速1.5ml/min。取供试品溶液20μl注入液相色谱仪,记录色谱图至主成分峰保留时间的两倍,面积归一化法计算纯度。
[0031]
本发明提供的方法具有以下优点:
[0032]
1、收率85%以上,纯度为99%以上,效果好;
[0033]
2、与现有方法(201811559536.1,公开号为cn111349016a,尤其是化合物c到d,以下简称2018年专利)相比,具有以下区别:
[0034][0035]
2018年专利化合物c到化合物d的反应式
[0036]
1)反应原料不同:2018年专利中的原料也用了2-氨基-5-氯吡啶,另一个原料反应部位是-coor,本发明是-cooh。
[0037]
2)缩合剂:2018年专利是胺酯交换反应,需要用到强碱,并且需要较高的反应温度,一般反应比较杂,收率不高没有使用缩合剂,本发明使用了两个缩合剂edci,hobt,没有使用强碱,并且缩合反应收率较高;
[0038]
3)反应温度,2018年专利反应温度为-20到130℃,本发明为20℃至有机溶剂的沸点。
附图说明
[0039]
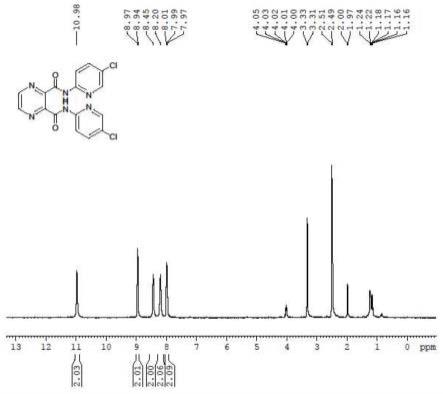
图1:吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的核磁共振氢谱(dmso-d6,400mhz)
[0040]
图2:吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的高分辨质谱(esi( ),m/z=389.0312);
[0041]
图3为吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的质谱图(局部放大)。
具体实施方案
[0042]
下述实施例是为了进一步说明本发明专利,并不构成对本发明的任何限制。
[0043]
实施例1:一种吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的合成方法
[0044]
将3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸(g)(5.57g,20mmol),三乙胺(4.04g,40mmol),edci(4.60g,24mmol),hobt(3.24g,24mmol)分散于二氯甲烷(50ml,为化合物g重量的8.98倍体积)中,搅拌10分钟。向反应混合物中加入2-氨基-5-氯吡啶(2.57g,20mmol),回流反应16小时。
[0045]
反应混合物逐渐降温至室温,加水搅拌,分液。水相以二氯甲烷萃取2次,有机相合并后,以饱和食盐水洗涤一次,硫酸镁干燥,过滤。滤液浓缩后经过柱层析(填充剂:200-300目硅胶,洗脱剂:石油醚
‑‑‑
石油醚/乙酸乙酯=10/1),得到类白色固体佐匹克隆杂质i吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺],收率87%,纯度99.5%。
[0046]
1h nmr(dmso-d6,400mhz)δ:10.981(s,2h),8.955(d,2h,j=9.6hz),8.466(s,2h),8.201(s,2h),8.226-8.268(m,2h),8.010-7.970(m,2h);m/z=389.0312(esi )。
[0047]
(见附图1-3)
[0048]
实施例2:一种吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺]的合成方法
[0049]
将3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸(化合物g)(5.57g,20mmol),三乙胺(4.04g,40mmol),edci(4.60g,24mmol),hobt(3.24g,24mmol)分散于dmf(50ml,为化合物g重量的8.98倍体积)中,搅拌10分钟。向反应混合物中加入2-氨基-5-氯吡啶(2.57g,20mmol),100℃反应16小时。
[0050]
反应混合物逐渐降温至室温,加水搅拌,再向水相中加入50ml二氯甲烷萃取两次,合并有机相,水相再以二氯甲烷萃取2次,有机相合并后以饱和食盐水洗涤一次,硫酸镁干燥,过滤。滤液浓缩后经过柱层析(填充剂:200-300目硅胶,洗脱剂:石油醚
‑‑‑
石油醚/乙酸乙酯=10/1)得到类白色固体佐匹克隆杂质i吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺],收率78%,纯度99.3%。
[0051]
方法同实施例1,区别是用dmf替换了二氯甲烷,但是用dmf做溶剂,副反应较多,所以收率略低。
[0052]
对比例1:
[0053]
将3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸(化合物g)(5.57g,20mmol),三乙胺(4.04g,40mmol),hatu(9.12g,24mmol)分散于二氯甲烷(50ml,为化合物g重量的8.98倍体积)中,搅拌10分钟。向反应混合物中加入2-氨基-5-氯吡啶(2.57g,20mmol),回流反应16小时。
[0054]
反应混合物逐渐降温至室温,加水搅拌,再向水相中加入50ml二氯甲烷萃取两次,合并有机相;水相再以二氯甲烷洗涤2次,有机相合并后以饱和食盐水洗涤一次,硫酸镁干燥,过滤。滤液浓缩后经过柱层析(填充剂:200-300目硅胶,洗脱剂:石油醚
‑‑‑
石油醚/乙酸乙酯=10/1)得到类白色固体佐匹克隆杂质i吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰
胺],收率55%,纯度92%。
[0055]
反应物中的催化剂用hatu(全名为2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯,也是一种常用的缩合剂,常用于氨酰键的合成)替换实施例1中的edci和hobt,其收率降低。
[0056]
对比例2
[0057]
将3-(5-氯吡啶-2-氨基甲酰)-2-吡嗪甲酸(化合物g)(5.57g,20mmol),2-氨基-5-氯吡啶(2.57g,20mmol)分散于无水二氯甲烷(50ml,为化合物g重量的8.98倍体积)中,冰水浴搅拌10分钟。向反应混合物中加入t3p(9.54g,30mmol),室温搅拌16小时。
[0058]
反应完成以后,加水搅拌,再向水相中加入50ml二氯甲烷萃取两次,合并有机相。水相以二氯甲烷洗涤2次。有机相合并后以饱和食盐水洗涤一次,硫酸镁干燥,过滤。滤液浓缩后经过柱层析(填充剂:200-300目硅胶,洗脱剂:石油醚
‑‑‑
石油醚/乙酸乙酯=10/1)得到类白色固体佐匹克隆杂质i吡嗪-2,3-二羧酸双[(5-氯-吡啶-2-基)-酰胺],收率25%,纯度92%。
[0059]
与实施例1的区别是,t3p是脱水剂,也用于缩合反应,当酸和胺缩合收率不高或者化合物对温度等条件敏感时会选用直接用它脱水,本品用它替换后,室温搅拌收率会降低;后期采用搅拌温度至回流,收率也没有提高。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。