基于mrna的肿瘤疫苗及其制备和联合抗癌方法
技术领域
1.本发明涉及生物医药技术领域,具体涉及一种基于mrna的肿瘤疫苗及其制备和联合抗癌方法。
背景技术:
2.本发明对于背景技术的描述属于与本发明相关的技术,仅仅是用于说明和便于理解本发明的
技术实现要素:
,不应理解为申请人明确认为或推定申请人认为是本发明在首次提出申请的申请日的现有技术。
3.通过改变或增强免疫系统抑制肿瘤发展的免疫治疗是目前肿瘤治疗的主流,为肿瘤治疗指明了新的方向。mrna疫苗已成为癌症免疫治疗的重要平台。mrna癌症疫苗因其高效能、安全给药、快速发展潜力和低成本生产而领先于其他传统疫苗平台。
4.癌症疫苗相对于传染病疫苗具有重大挑战。首先,大多数传染病疫苗是预防性的,而癌症疫苗是治疗性的。预防性癌症疫苗的案例非常罕见,只有两种fda批准的疫苗,这两种疫苗用于预防病毒诱导的恶性肿瘤(hpv和hbv)。其次,大多数传染性疾病的抗原(细菌或病毒驱动)是典型的由mhcii分子呈现的外源性基序。针对这些外源性抗原的疫苗诱导中和性抗体介导的体液反应,在某些情况下,cd4 t细胞介导的免疫应答是部分参与和必需的,而cd8 细胞毒性t细胞在清除体细胞突变的恶性细胞中发挥关键作用。因此,抗肿瘤治疗疫苗不仅需要增强体液反应、cd4 t细胞反应,还需要激活mhci介导的cd8 t细胞反应,这进一步增加了有效增强抗肿瘤免疫的难度。
5.高效递送高度免疫原性肿瘤特异性抗原。识别和高效递送高度免疫原性肿瘤特异性抗原是高效抗癌疫苗开发的另一主要障碍。肿瘤抗原在不同的个体之间高度可变,有些免疫原性较差,可以侵入宿主的免疫识别系统;对于具备免疫原性的抗原,抑制性的微环境也可以防止有效的t细胞浸润并导致t细胞衰竭;针对治疗性疫苗,需要使用较预防性疫苗更大剂量的多次/可重复剂量,以提高mrna和携带者的安全性标准。在其他癌症疫苗中,包括基于树突状细胞(dc)疫苗和基于蛋白质的疫苗,信使rna脱颖而出有以下几个原因:(1)mrna可同时编码多种抗原,或同时具有mhci和mhcii结合表位的全蛋白,促进体液和细胞适应性免疫反应,增强抗肿瘤免疫。(2)与dna疫苗相比,mrna疫苗是非整合的,可降解性强,无插入突变潜能;与蛋白质或细胞介导的疫苗相比,ivt生产的mrna不含细胞和致病性病毒成分,没有感染的可能性。因此,mrna是一个功能强大、用途广泛的癌症疫苗平台,其临床转化的成功发展将显著增强人类对抗癌症的能力。利用mrna的内在固有免疫,通过设计先进和可耐受的传递系统来提高抗原表达和呈现的效率,以及修改mrna结构以延长和控制表达时间成为现阶段主要面临的技术问题。
6.mrna编码新抗原引领个性化疫苗发展。新抗原成为癌症疫苗(cvs)的首选靶点,尤其包括高通量检测突变相关表位的方法,以及预测新表位的生物信息工具,均获得良好效果。与本发明相关的肿瘤特异性抗原(taa/tsa)和新抗原及治疗方法主要包括:
①
人表皮生长因子受体(her2)促进细胞转化和肿瘤生长。该受体的过表达与不良的临床结果相关,导
致侵袭性肿瘤表型和生存期降低。因此,该受体作为肿瘤相关抗原(tumor-associated antigen,taa)已成为癌症治疗的重要靶点。egfr与肿瘤细胞的增殖、肿瘤侵袭、转移及细胞凋亡的抑制有关。在癌症中,变异的egfr得到持续性的激活,从而引起细胞无节制的扩增并最终导致癌症。因此,egfr是一个癌症特异性靶点。
②
b7-h3是一种来自b7家族的新型免疫检查点。靶向pd-1、ctla4等免疫疗法的成功,也为研究者开发以b7-h3为靶点的新免疫疗法提供了范例和方向。然而,b7-h3受体的未知特性极大地阻碍了b7-h3拮抗剂的开发。提高对b7-h3介导的肿瘤发生调控分子过程的理解,在新的免疫检查点b7-h3的基础上研究不同器官的细胞治疗成为现有技术亟需解决的技术问题。使用单克隆抗体阻断免疫检查点,ctla-4、程序性细胞死亡蛋白1(pd-1)和pd-1配体1(pd-l1)在各种癌症患者中显示出显著的临床成功。这种经验也可以应用到b7-h3。然而,由于缺乏人类b7-h3特异性阻断单克隆抗体,这一策略的临床应用受到了阻碍。通过不同免疫疗法的联合治疗来提高患者的治疗效果和生存率,多种免疫检查点抑制剂的联合疗法成为亟需解决的技术问题。
③
新抗原tenm4具有重要作用。与正常组织相比人肺腺癌、低级别胶质瘤和胰腺腺癌中tenm4 mrna的表达显著增加,在肝癌、子宫内膜癌、胃癌、肾癌中tenm4的表达与预后呈相反的相关趋势,tenm4表达的升高与患者生存期的降低相关。由于tenm4是一种跨膜蛋白,在发育过程中表达受到限制,且在少数成人组织中发现低表达,因此被认为是药物和免疫治疗的良好靶点。因此,实现编码tenm4的疫苗的开发将促进持久的体液免疫和细胞反应的产生,以抑制肿瘤进展,推动该编码疫苗的开发成为现有技术亟需解决的技术问题。
④
tenm4在乳腺癌发展和治疗中具有重要价值。乳腺癌的治疗是基于er、pr或her2受体的表达,靶向治疗在治疗某些乳腺癌亚型方面取得了相当大的成功。然而,tnbc是最具侵袭性的亚型之一,目前仍没有真正有效的靶向治疗方法。此外,虽然原发性乳腺癌是高度可治疗的(80%-99%的被诊断为i/ii期乳腺癌的女性可存活到5年)。tenm4具有良好的肿瘤相关抗原的特点,成为乳腺癌及其他相关癌症预防及治疗性疫苗成为亟需解决的技术问题。
7.癌症疫苗的治疗效果取决于几个因素,包括:(i)选择最合适的目标肿瘤抗原(ii)对抗免疫抑制肿瘤微环境的能力,以及(iii)疫苗接种平台在诱导强大细胞免疫方面的效率。在抗原选择方面,新抗原(nag)正成为癌症疫苗的一个有吸引力的目标。目前没有一种单一的方法可以可靠地识别免疫原性(有效的新抗原),这使得以往的经验判断在个性化接种的临床环境中仍然不可行。
8.car-t细胞治疗及car分子优化具有重要作用,大多数car-t治疗的成功是在血液癌中。当面对实体肿瘤时,car-t细胞的治疗效果有限。主要是由于在免疫抑制肿瘤微环境(tme)中难以归位、浸润和生存,以及肿瘤抗原逃逸和异质性。此外,car-t治疗相关的副作用也有报道,包括靶向非肿瘤毒性、神经毒性、细胞因子释放综合征(crs)等。
9.基于上述技术现状,联合治疗成为重要的技术手段。然而,α-pd-1/pd-l1治疗的低反应率仍有待解决。对于大多数癌症患者来说,pd-1/pd-l1通路并不是抗肿瘤免疫的唯一限速因子,通过阻断pd-1/pd-l1轴不足以激发有效的抗肿瘤免疫应答。虽然α-pd-1/pd-l1治疗在某些患者中显示出强有力的抗肿瘤作用,但由于原发性或获得性治疗的耐药性,大多数患者无法从α-pd-1/pd-l1治疗中获益。对于无应答者,pd-1信号并不是癌症免疫周期的限速变阻器,通过阻断pd-1或pd-l1不足以恢复抗肿瘤免疫。另外,car-t细胞治疗在大多数实体肿瘤中的疗效并不明显,这部分归因于免疫抑制tme。尽管有几十种联合方案在临床
前研究中显示出强有力的抗肿瘤活性,但一些积极的临床前发现不能在临床中得到验证。目前,只有α-pd-1/pd-l1联合化疗、血管生成抑制剂或α-ctla-4获得fda或nmpa的批准。对于大多数组合,显著的抗肿瘤活性是有限的。
10.因此,如何选择一个最佳的临床前模型是一个巨大的挑战,以确定联合方案的活性。相对于广泛应用的同基因小鼠模型,人性化患者来源模型可以提供更宝贵的疗效评价。此外,不适当的联合治疗将使患者暴露于明显更高的毒性。如何优化给药方案,包括剂量、时间和顺序,是联合治疗发展的另一个挑战。最后,还不清楚如何选择合适的联合治疗和寻找生物标志物预测治疗反应。结合肿瘤的异质性和演化,动态监测tme的免疫检测,为指导精准免疫治疗提供了实时的生物标志物。另外,mrna疫苗的应用还存在由于不稳定性、先天免疫原性和体内传递效率低下而受到限制等缺点。
发明内容
11.鉴于解决现有技术肿瘤治疗中的疫苗乏力、免疫逃避等问题,mrna癌症疫苗相对于传染病疫苗具有重大挑战的缺陷,本发明的目的是提供一种基于mrna的癌症预防和治疗疫苗,免疫检查点疫苗,重定向car-t细胞和免疫佐剂的制备及抗肿瘤免疫联合治疗方法。
12.第一方面,本发明提出了一种基于mrna的肿瘤疫苗;进一步地,所述疫苗包括预防和治疗的疫苗;所述疫苗还包括免疫检查点疫苗。
13.第二方面,本发明所述疫苗包括car-t细胞及免疫佐剂的mrna表达体及其脂质递送系统;进一步地,mrna表达体以嵌合抗原的形式表达肿瘤特异性抗原、突变抗原、新抗原及car-t细胞受体和免疫佐剂;进一步地,所述特异性抗原首选包括但不限于her2/neu,her2/neu和sars-cov-2野生或变体rbd抗原,新抗原tenm4,免疫检查点b7-h3,双特异性b7-h3嵌合抗原受体car分子及免疫佐剂il-15;进一步地,所述脂质递送系统包括脂质体和脂质纳米颗粒系统。
14.第三方面,本发明提出了一种基于mrna肿瘤疫苗的联合抗癌方法;进一步地,本发明包括但不限于预防、治疗疫苗、免疫检查点疫苗或抗体、car-t细胞和佐剂的合理适当配伍治疗。
15.为了实现上述目的,本发明所采取的技术方案为:提出一种基于mrna的肿瘤疫苗,包括mrna表达体、肿瘤特异性基因及胞内递送系统;所述mrna表达体中,mrna为自扩增rna,包括编码抗原和rna复制酶,扩增重组mrna。
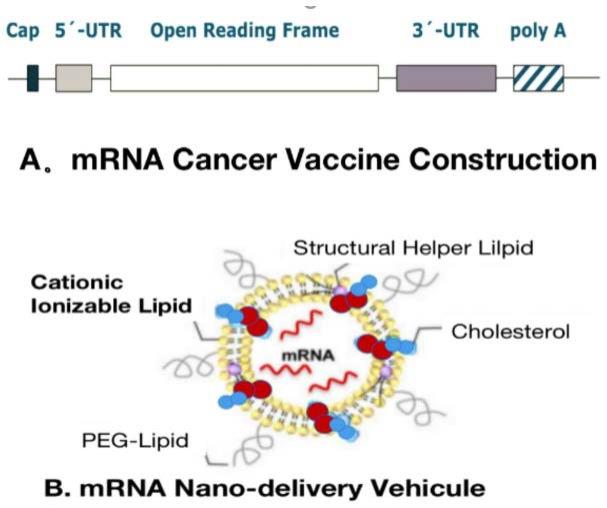
16.进一步地,自扩增rna包括体外转录mrna,由抗原编码的开放阅读框orf、5’和3’端未翻译区utr、一个7甲基鸟苷5’帽结构、合并到第一个核苷酸的序列和3’端多聚腺苷酸尾部;所述的表达体还包括结构调整,密码子优化及核苷酸修饰。
17.进一步地,肿瘤特异性基因包括野生型,野生型 多表位肽抗原,及野生型 sars-cov-2结合阈rbd野生或变异体肽抗原;所述的肿瘤特异性基因包括但不限于her2/neu mrna和tenm4 mrna;优选地,所述肿瘤特异性基因为her2/neu mrna。
18.进一步地,肿瘤疫苗诱导细胞产生肿瘤特异性抗原,所述肿瘤特异性抗原包括肿瘤组织特异性基因表达产物,基因突变,尤其是通过高通量生物技术筛选的特异性新基因;还包括所述基因及其所选的多表位抗原;所述的基因多表位抗原辅助于肿瘤特异性抗原或新抗原,其多表位肽分子包含2-200个氨基酸,多表位肽分子的数量为2-200个抗原分子。
19.进一步地,肿瘤疫苗为预防和/或治疗性的。
20.进一步地,疫苗为免疫检查点疫苗,所述免疫检查点疫苗所涉基因包括但不限于b7-h3 mrna。
21.进一步地,肿瘤疫苗还包括双特异性双功能car-t分子,所述双特异性双功能car-t分子为高通量筛选的细胞治疗性优选基因,包括但不限于b7-h3嵌合受体;优选地,设计利用双特异性t细胞参与bites作为car-t和肿瘤细胞之间的适配器的car-t;优选地,设计基于cars分子的基于蛋白开关的逻辑门;包括但不限于帮助car-t细胞对抗肿瘤免疫抑制微环境tme的优选方法,包括细胞因子、趋化因子受体、显性负性受体、肝素酶和crispr介导的基因组编辑和t细胞文库筛选的表达;优选地,除car-t细胞外,还包括工程化和优化的car-nk细胞和car-m细胞或树突状细胞。
22.进一步地,肿瘤疫苗还包括胞内递送系统;优选地,所述胞内递送系统为脂质递送系统,所述脂质递送系统配方包括可电离的阳离子脂质、磷脂、胆固醇和脂质锚定的聚乙二醇;优选地,lnp配方包括rna-脂质复合物、提供结构刚性的脂质以及能够修饰颗粒表面特性的脂质化聚合物涂层。
23.进一步地,肿瘤疫苗还包括免疫佐剂;所述免疫佐剂为编码免疫刺激蛋白的mrna,所述mrna包括gs-csf,ifn-gamma、il-12、il32、ox40l、cd40l、cd70;优选地,包括但不限于il-15。
24.进一步地,还提出一种基于mrna疫苗的联合抗癌治疗方法,包括预防、治疗疫苗、免疫检查点疫苗或抗体、car-t细胞或car-nk细胞和免疫刺激佐剂的合理适当配伍治疗;所述的免疫检查点抑制剂抗体,包括但不限于pd-1/pd-l1。
25.本发明实施例具有如下有益效果:以达到全面全方位多基因多因素的精准肿瘤免疫治疗效果。
附图说明
26.图1为本发明一实施例中mrna癌症疫苗表达体的分子结构特征和脂质纳米包裹递送系统图;
27.图2为本发明一实施例中b7-h3/cd3 bite及b7-h3重定向car-t细胞结构示意图;
28.图3为本发明一实施例中b7-h3/cd3 bite和b7-h3重定向car-t细胞的免疫活性检测图;
29.图4为本发明一实施例中b7-h3/cd3 bite和重定向car-t细胞体外诱导的t细胞介导细胞毒性检测图;
30.图5为本发明一实施例中b7-h3/cd3 bite和b7-h3重定向car-t细胞联合进行模型动物体内抗肿瘤杀伤实验图;
31.图6为本发明一实施例中mrna癌症疫苗构建及其t细胞免疫应答检测图。
32.图7为本发明一实施例中早期接种预防性疫苗,预防肿瘤生长的检测图。
33.图8为本发明一实施例中转移肿瘤模型早中期治疗疫苗干预可控肿瘤生长(早中期疫苗治疗)检测图。
34.图9为本发明一实施例中对高负荷肿瘤的治疗疫苗干预,即对晚期肿瘤的疫苗干预效果检测图。
35.图10为本发明一实施例中抗肿瘤的免疫联合治疗效果检测图。
具体实施方式
36.下面结合实施例对本技术进行进一步的介绍。
37.为了更清楚地说明本发明实施例或现有技术中的技术方案,在下述说明中,不同的“一实施例”或“实施例”指的不一定是同一实施例。不同实施例之间可以替换或者合并组合,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些实施例获得其他的实施方式。
38.通过改变或增强免疫系统抑制肿瘤发展的免疫治疗是目前肿瘤治疗的主流方向,mrna疫苗已成为癌症免疫治疗的重要平台。在接种过程中,裸或载药的mrna疫苗在抗原提呈细胞(antigen presenting cell,apcs)中有效表达肿瘤抗原,促进apc激活和先天/适应性免疫刺激。mrna癌症疫苗因其高效能、安全给药、快速发展潜力和低成本生产而领先于其他传统疫苗平台。
39.本发明研究了适当的mrna结构修改(如密码子优化、核苷酸修改、自扩增mrna等)和脂质递送系统配方方法(如脂质纳米粒(lnps)、聚合物、多肽等);调整给药途径,将多种mrna疫苗与其它免疫治疗药物(如检查点抑制剂)共给药,进一步增强了宿主的抗肿瘤免疫力,并增加了肿瘤细胞被消灭的可能性。随着mrna癌症疫苗在针对多种侵袭性实体肿瘤的几个临床试验中取得了有益的治疗效果,本发明将自身研究成果结合mrna疫苗的最新进展、最新的癌症抗原和肿瘤微环境的改善等,提出了mrna癌症疫苗及其制备和联合抗癌的新方法。
40.本发明首先提供了各种疫苗及成分表达体的设计生产制备过程。
41.(1)构建各种疫苗及car-t细胞car分子和免疫刺激因子的表达质粒载体
42.本发明在野生和变异型肿瘤多表位抗原和突变序列(见序列表seq id no:1~6)的基础上,进行密码子优化和质粒构建。mrna疫苗和表达体的分子结构特征见图1。本发明利用包含每个所需抗原及突变的寡核苷酸进行多重pcr片段扩增,并利用重叠片段组装生成每个疫苗和表达体的完整序列。进一步地,本发明在前述密码子优化基础上,对其抗原mrna表达质粒优化和进行核苷酸修饰,在此背景下获得相关突变。该质粒含有c-末端18个氨基酸的缺失,本发明在前期相关实验证明这些缺失可以导致更高的假病毒滴度。另外,本发明进行rna自扩增设计和mrna加帽一步完成设计。利用in-fusion hd克隆试剂盒(takara)将组装好的片段插入noti/xbai消化后的ptwist-cmv-betagloin-wpre-neo慢病毒表达载体中。研究中使用的所有质粒dna均经过全质粒深度测序(illumina)验证,以确认仅存在预期的突变。
43.另外,所选的疫苗抗原表达序列中,预防疫苗以her2/neu为首选,主要由于egfr是一个在肺癌等多种实体瘤细胞中表达或异常表达,而在普通细胞中表达很微弱的细胞表面抗原。egfr与肿瘤细胞的增殖、肿瘤侵袭、转移及细胞凋亡的抑制有关。在癌症中,变异的egfr得到持续性的激活,从而引起细胞无节制的扩增并最终导致癌症。因此,egfr是一个癌症特异性靶点。
44.研究发现,约30%的转移性乳癌已显示过表达her2/neu。这种过表达与乳癌患者的预后差相关。已在人非小细胞肺癌(nsclc)中鉴定了egfr突变,5%的人肺鳞状细胞癌具
有egfrviii突变以及10-30%的肺腺癌具有egfr激酶结构域突变。当t790m和c797s同时突变时,t790m和c797s位于同一条染色体上,这种构型称为顺式(为主要类型,占85%);t790m和c797s位于不同染色体上,这种构型称为反式,egfrt790m突变占40%~60%。30%~40%会继发egfr c797s突变。目前研究结果表明,t790m、c797s突变及不同构型与肿瘤患者的耐药性、疗效预测、预后极其相关。本发明增加了突变位点的多表位肽抗原。
45.b7-h3是一种来自b7家族的新型免疫检查点。由于其在肿瘤组织中过表达而在正常组织中表达有限,并参与肿瘤微环境(tumor microenvironment,tme)的形成和发展,是一个有吸引力和前景的肿瘤免疫治疗靶点。b7-h3作为癌症免疫治疗靶点的前景已经刺激了b7-h3靶向治疗策略的进展。与其他免疫检查点相比,b7-h3不仅影响先天免疫和适应性免疫,而且还通过各种非免疫途径调节癌细胞的侵袭性。各种抗b7-h3途径已经在临床前和临床试验中进行了研究,并证明了其临床应用的可行性。b7-h3在各种癌症中得到了广泛的研究,包括但不限于乳腺癌、肺癌、卵巢癌、脑瘤、胃癌、鳞状细胞癌。它的存在与预后恶化、生存不良和复发率有关。现有研究评估了b7检查点分子在卵巢癌(ovca)中的表达,发现b7-h3高表达,而不是pd-l1。目前的多项研究表明,b7-h3可能通过抑制活化t细胞的核因子nf-κb和激活蛋白1介导的信号通路,抑制cd4 和cd8 t细胞的增殖,并减少il-2和ifn-γ的产生。b7-h3除抑制t细胞外,还抑制nk细胞活性。b7-h3形成的抑制性免疫微环境有助于癌症避免免疫破坏。b7-h3的异常表达不仅是一种生物标志物,也是多种癌症免疫治疗方法的一个有前途的免疫检查点靶点,特别是在癌症启动细胞(cics)上的表达。
46.本发明特别强调,由于b7-h3在正常组织很少表达,故本发明构建b7-h3免疫检查点疫苗,通过自身组织产生抗体,以获得有效的免疫检查点抑制效果;但它同时也是一个预防和治疗疫苗。
47.tenm4突变和重排最近在许多肿瘤中被发现。肿瘤中观察到的tenm4突变大部分发生在乳腺癌中,其中tenm4在细胞迁移和干细胞中发挥作用。由于tenm4是一种跨膜蛋白,在发育过程中表达受到限制,且在少数成人组织中发现低表达,因此可能被认为是药物和免疫治疗的良好靶点。它在分化的上皮细胞和未分化的癌症干细胞(csc)中的表达意味着它的靶向可能有利于限制肿瘤负荷,但最重要的是,可以消除csc,从而限制肿瘤细胞的传播。编码tenm4的疫苗的开发将促进持久的体液免疫和细胞反应的产生,从而抵消乳腺肿瘤的进展。tenm4通过抗癌疫苗接种,可以保证其安全性。所以,本发明首选tenm4作为乳腺癌的预防和治疗性疫苗,包括但不限于乳腺癌的预防和治疗。
48.⑵
mrna的体外转录(ivt)及纯化
49.用于生成mrna的体外转录(ivt)酶促反应依赖于t7、sp6或t3rna聚合酶催化从相应的dna模板合成靶mrna。通过纯化质粒的线性化或通过pcr扩增感兴趣的区域。除了线性dna模板外,rna聚合酶、核苷酸三磷酸(ntps)底物、聚合酶辅因子mgcl2、含有多胺和抗氧化剂加入ph缓冲液中完成,反应只需几个小时。此外,这种缩短的时间降低了污染发生的可能性。一般来说,每毫升反应可以得到毫克的mrna。此外,由于其不依赖于编码在模板中的抗原,生产过程可以标准化。
50.mrna的纯化:在实验室规模的mrna纯化过程包括dnase i酶切,然后licl沉淀。更大规模的净化是使用成熟的色谱策略结合切向流动过滤获得。另外,新型色谱法也可用来补充标准的纯化方法。由于相关反应杂质降低了翻译效率并修改了免疫刺激谱,因此,去除
这些产物相关的反应杂质对mrna的性能至关重要。当核苷修饰的mrna被反相高效液相色谱(reverse phase hplc)纯化后,本发明观察到蛋白质产量增加了数十倍。
51.(3)脂质纳米递送系统包裹及免疫佐剂
52.由阳离子脂质1,2-二油酰-3-三甲基丙烯铵(dotap)、一种辅助脂质1,2-二硬脂酰-sn甘油-3-磷酸胆碱(dspc)]、胆固醇和聚乙二醇(peg)-脂质[(2-[(聚乙二醇)2000]-n,n-二十二烷基乙酰胺(peg2000-dma)或1,2-二肉豆荚酰-丙三醇-甲氧基聚乙二醇-2000(peg2000-dmg)]组成的脂质纳米递送系统,使纳米颗粒具有水合层,提高胶体稳定性,减少蛋白质吸收。
[0053]
其脂质体积/重量比例为:dotap:dspc:胆固醇:peg200-dma=10-15:1:1-2:3-5。脂质纳米颗粒为100-300nm。
[0054]
mrna疫苗的脂质系统配方通常包含可电离的阳离子脂质、磷脂、胆固醇和脂质锚定的聚乙二醇(peg),dlin-mc3-dma,也被称为mc3,是为sirna开发的一种可离子化氨基脂,最近被用于mrna疫苗递送的lnps。磷脂是已知的促进膜的形成和破坏过程,促进内体逃逸。可电离脂质在生理ph值下为中性,避免了循环中的任何阳离子电荷,在ph 6.5时在内体中被质子化,从而促进核内体释放。可电离的脂质复合物与mrna形成核心结构,而辅助脂质(如磷脂、胆固醇和/或peg脂质)包裹脂质信使rna复合物。反过来,peg-脂质保护纳米颗粒外壳。因此,胆固醇在lnps中起着稳定因素的作用,在细胞转染中起着至关重要的作用。脂锚定的peg优先沉积在lnp表面,使其在空间上稳定,也减少了与蛋白的非特异性结合。此外,较高的peg含量可以减少lnps对血浆蛋白的特异性吸收,减少细胞摄取以及与核内体膜的相互作用,从而增加lnps的血液循环时间,提高mrna的传递效率。
[0055]
脂质纳米颗粒可以保护mrna免受酶的攻击,并提高细胞对mrna的摄取和表达,比裸体mrna提高1000倍。可离子化脂质,在生理ph条件下具有中性到温和的阳离子电荷,从而减少非特异性脂质-蛋白质相互作用,促进核酸在细胞质中释放。与脂类相比,这种特性具有永久性的降低毒性和延长血液循环寿命的优点。其他脂质被认为是“辅助脂质”。因为它们可能会影响复合物lnp-mrnas的结构特性,以提高其稳定性或促进其胞内摄取和胞质进入。因此,典型的lnp配方包括rna-脂质复合物、提供结构刚性的脂质以及能够修饰颗粒表面特性的脂质化聚合物涂层。例如,由阳离子脂质1,2-二油酰-3-三甲基丙烯铵(dotap)和辅助脂质1,2-二油酰-sn甘油-3-磷酸乙醇胺(dope)与mrna结合而成的阳离子脂质体包裹mrna疫苗。
[0056]
免疫佐剂是一类编码免疫刺激蛋白的mrna,如gs-csf,ifn-gamma、il-12、il32、ox40l、cd40l、cd70等,包括但不限于il-15;该佐剂可以刺激cd8 t细胞和nk细胞增殖,增加肿瘤细胞毒性;可以保留低分化的干细胞记忆(tscm)表型,减少衰竭,增强抗原结合时的增殖。
[0057]
(4)mrna疫苗连续生产的微流体方法
[0058]
mrna疫苗精确、安全、灵活,易于大规模生产,可用于临床级应用。尽管mrna的ivt反应比大多数已建立的疫苗生产更安全、更快,但它依赖于使用昂贵和有限的材料。疫苗的下游加工仍然不完善,而且依赖于缺乏可扩展性和成本效益的方法。将这一过程转变为连续生产可以克服这些瓶颈。本发明提出一个微流体方法原位酶反应加上产品去除处(ispr)、底物上料和监控模块及多通道使用色谱法取代使用多个色谱步骤,该新的连续生
产方法,结合高通量纯化和定义明确的分析方法的化合物再循环,可以实现可持续、灵活和成本效益高的疫苗生产,从而实现按需定制和规模化生产。
[0059]
⑸
多表位抗原疫苗与新抗原鉴定
[0060]
多表位疫苗可能是一种解决生物信息学工具预测新抗原不准确性的方法。目前,还没有一种方法能够准确可靠地预测和鉴定新抗原。解决这个问题的一种方法是设计多表位疫苗。使用一个可以容纳大量新抗原的平台,可能有助于克服预测算法的局限性。在一项研究中,从小鼠ct26结肠癌细胞系中选择的迄今为止用于新抗原疫苗的最大数量的多表位肽抗原,结合针对31个新抗原的腺病毒疫苗治疗,产生了强有力的抗肿瘤免疫反应,产生了超过1000个抗原特异性ifn-γ分泌淋巴细胞/百万脾细胞,产生了cd8 和cd4 t淋巴细胞,增加了完全消退小鼠的数量(50%)。另一个选择是扩大本发明对潜在新抗原的分析,包括其他类型的潜在免疫原性改变。
[0061]
本发明选择临床癌细胞系或直接从患者身上取得的癌组织,通过下一代测序(ngs)进行dna和rna测序。候选新抗原(nag)是从确定的突变中选择的,其基于:(i)、mhc-i和ii预测的结合亲和力;(ii)、肿瘤dna中的突变等位基因频率;(iii)、mrna表达程度。并根据(i)mhc class i(预测ic50≤500nm)和mhc class ii(结合评分≤1)结合预测,(ii)肿瘤等位基因频率(maf≥25%)和(iii)rna表达(≥1突变rna)选择nag。
[0062]
优先根据单核苷酸变异(snv)突变,以及基于野生型特定突变的每个氨基酸(aa)上游和下游25-50aa的总长度,以最大数量生成一个包含cd4 和/或cd8 t细胞表位的抗原肽。利用mrna表达体克隆能力强的特点,将若干个nag首尾相连合成一个人工蛋白。通过体外ifn-γ酶联免疫吸附试验(单次注射该载体可诱导小鼠有效t细胞免疫),预测新抗原中能够引起可检测到的免疫应答个数,通过细胞内细胞因子染色(ics)和荧光激活细胞分选(facs)分析免疫原性nag个数,确定单个nag诱导的t细胞应答的质量
[0063]
本发明在前期研究的基础上,认为多表位疫苗最关键的应该是对野生型抗原肽的强烈交叉反应,这样才能在肿瘤的防治中发挥作用。gad-ct26-31肽研究的致命缺点在于对野生型多肽的无交叉反应,说明未来可能在临床实践中对原发野生型肿瘤缺乏强力的免疫性。其原因可能在于多表位抗原肽的免疫原性不够,不足以产生针对野生型多肽的强烈交叉免疫反应。
[0064]
因此,本发明认为多表位抗原的质量很重要,即与野生型结合位点的匹配,特别是产生的抗体和免疫细胞与之结合的交叉结合能力很重要,而不在于多表位肽的数量很多。上述31个预测的新抗原中有7个能够引起可检测到的免疫应答,有效的多表位抗原少于25%。因此,本发明在权利要求中提出多表位肽的长度应在2-200氨基酸之间,以30-100个氨基酸为适当;以能够产生足够的交叉免疫反应为基础。特别是,多表位抗原起辅助作用,而应以全长序列的抗原为中心,除非所选的多表位肽序列均为突变有价值的免疫刺激基序。
[0065]
本发明所述多表位抗原,不是纯粹多表位抗原疫苗,特别强调在预防性疫苗中,以人体组织不存在的多表位抗原为主,在治疗疫苗中,以自身个体化肿瘤特异性抗原为主。这样,既达到预防和治疗的目的,又很少引起非特异性的副反应,减少疫苗治疗的副作用。
[0066]
⑹
car-t分子的设计与优化
[0067]
嵌合抗原受体(car)t细胞治疗在过去十年中已经发展成为最有前途的癌症治疗
方法之一,特别是对血液肿瘤。car-t治疗包括使用car分子对患者的t细胞进行体外基因工程,使t细胞具有针对目标肿瘤细胞的定向特异性,并随后将car-t细胞注入患者体内进行癌症治疗
[0068]
第一代car,通常包含用于抗原识别的细胞外单链可变片段(scfv),用于信号转导的铰链(h)和跨膜(tm)域,以及用于激活的细胞内cd3z。第二代car-t细胞在跨膜和cd3z结构域之间添加了共刺激结构域(例如cd28),并被证明可以解决第一代car-t细胞相关的低增殖问题。第三代car,其特征是跨膜和cd3z结构域之间合并了两个共刺激结构域(如cd28和ox40),也被开发并显示进一步增强car-t细胞的性能。
[0069]
大多数car-t治疗的成功是在血液癌中。当面对实体肿瘤时,car-t细胞的治疗效果有限,主要是由于在免疫抑制肿瘤微环境(tme)中难以归位、浸润和生存,以及肿瘤抗原逃逸和异质性。此外,car-t治疗相关的副作用也有报道,包括靶向非肿瘤毒性、神经毒性、细胞因子释放综合征(crs)等。
[0070]
因此,car-t细胞工程目标是(a)提高car-t细胞的疗效,以克服关于car-t治疗在实体肿瘤中的无效问题,(b)提高car-t细胞的安全性,以减轻和/或最小化与以前的car-t产品相关的不良毒性。本发明在这些方面已经做出了巨大的努力。在这里,本发明提供了一个关于car-t细胞设计的新策略,旨在提高car-t治疗的有效性和安全性
[0071]
设计car新分子:(a)car分子的外结构域、铰链结构域(h)、跨膜结构域(tm)和内结构域的修饰。(b)双特异性t细胞参与(bites)设计,或三特异性杀手接合器(trikes)。(c)设计利用双特异性t细胞参与(bites)作为car-t和肿瘤细胞之间的适配器的car-t。(d)设计car实现基于蛋白开关的逻辑门,以整合来自两个或更多肿瘤抗原的输入。
[0072]
通过修饰非car分子来改造装甲car-t细胞。(a)car-t细胞面对实体肿瘤的免疫抑制tme。(b)旨在帮助car-t细胞对抗免疫抑制tme的代表性方法,包括细胞因子、趋化因子受体、显性负性受体、肝素酶和crispr介导的基因组编辑和t细胞文库筛选的表达。
[0073]
由各种外部刺激控制car-t细胞的on和off开关,如肿瘤抗原、小分子、光、超声波和其他环境线索,可用来激活功能性car表达(on-switch)或失活car表达t细胞(off-switch)。
[0074]
自动化的car-t细胞基于合成的notch受体(synnotch)系统的and-gate t细胞工程,可以精确控制car-t细胞信号通路。为可控系统量身定制的基于crispr的人类t细胞高通量筛选,可能会改变car的工程方向和结果。
[0075]
细胞因子分泌重组白细胞介素(il)-12已被临床应用于多种类型的实体肿瘤。本发明用il-15培养的car-t细胞可以保留低分化的干细胞记忆(tscm)表型,减少衰竭,增强抗原结合时的增殖。事实上,包括巨噬细胞和树突状细胞在内的多种细胞类型都可以产生il-15,il-15可以刺激cd8 t细胞和nk细胞增殖,增加肿瘤细胞清除。
[0076]
本发明用高通量的生物技术方法筛选得到的最有力地提高细胞治疗性能的候选基因b7-h3嵌合受体,已被证明可以增强t细胞的适应性,促进关键效应因子的表达,并改善体内实体肿瘤清除。
[0077]
含有两个串联配体结合域的双特异性car已被证明在car-t信号转导中有效地实现or逻辑门,双特异性car对靶向癌细胞的亲和力增强,可以增强免疫治疗,并有助于避免异质性肿瘤可能发生的抗原逃逸。
[0078]
car-nk具有毒性小、显著降低gvhd风险的优点,是一种更安全、更普遍的治疗方法。它的非抗原杀伤能力也可以用来解决某些肿瘤中抗原逃逸的问题。然而,car-nk在基因传递和细胞扩张方面也有困难。car-m和car-nk作为car-t治疗的替代和补充治疗还需要更多的优化。
[0079]
⑺
mrna疫苗联合抗癌方法
[0080]
虽然α-pd-l1抗体(atezolizumab,durvalumab,avelumab)已被批准用于各种类型的癌症。然而,α-pd-1/pd-l1治疗的低反应率仍有待解决。对于大多数癌症患者来说,pd-1/pd-l1通路并不是抗肿瘤免疫的唯一限速因子,通过阻断pd-1/pd-l1轴不足以激发有效的抗肿瘤免疫应答。
[0081]
目前,一些联合疗法已经被证实,包括α-pd-1/pd-l1加化疗、放疗、血管生成抑制剂、靶向治疗、其他免疫检查点抑制剂、共刺激分子激动剂、干扰素基因刺激剂激动剂、粪便微生物群移植、表观遗传调节剂、或代谢调节剂,具有优越的抗肿瘤功效和更高的反应率。此外,含有α-pd-1/pd-l1部分的双功能或双特异性抗体也具有更强的抗肿瘤活性。这些组合策略同时促进癌症免疫周期的多个过程,消除免疫抑制剂的不足。
[0082]
本发明特别强调预防疫苗、治疗疫苗、免疫检查点疫苗、car-t细胞与免疫佐剂的联合治疗,但不限于此,如sting激动剂在促进免疫细胞浸润,增强apc、nk和t细胞的功能中的应用等;从而获得全面和全方位的精准免疫治疗效果。
[0083]
其中,与本发明相关的肿瘤特异性抗原(taa/tsa)和新抗原及治疗方法主要包括:
[0084]
(1)人表皮生长因子受体(her2)促进细胞转化和肿瘤生长。女性死亡的主要原因之一是乳腺癌,包括过表达her-2/neu(erbb2)的分子亚型。her-2/neu最早于1985年在人乳腺癌细胞mac117中被发现,有5-10倍的受体过表达。her-2/neu是一种185kda的跨膜蛋白,与her1(egfr)、her3和her4共同属于her蛋白家族。许多表皮生长因子(egf)配体已经被定义,但没有一个是her2特异性的。当配体与her1、her3或her4结合时,受体二聚化发生,her2成为首选的二聚体伴侣;随后,酪氨酸激酶胞内结构域的激活触发不同的下游通路。当her2作为一种癌基因发挥作用时,通过基因的过表达而导致her2更容易异源二聚,进而促进细胞转化和肿瘤生长。在缺乏适当治疗的情况下,her2高表达的乳腺癌(bc)与侵袭性的临床行为相关。在her2阳性的bcs中,已经观察到t细胞免疫和癌症复发率降低之间有很强的相关性。肿瘤浸润淋巴细胞(tumor infiltrating lymphocyte,tils)每增加一个百分比单位,复发风险相对降低3%。此外,her2阳性肿瘤细胞与肿瘤微环境(tme)中潜在配体相互作用的特异性被认为促进了针对her2蛋白的免疫应答。准确地说,针对her2阳性bc的免疫应答是由细胞毒性t淋巴细胞(cd8 )和cd4 t细胞诱导的。细胞毒性t淋巴细胞识别主要组织相容性复合体(mhc)i类分子呈现的her2多肽,诱导细胞周期阻滞、凋亡和干扰素γ(ifn-γ)介导的杀伤肿瘤细胞;cd4 t细胞和抗her2抗体与更强的抗her2免疫反应相关。鉴于上述原因,现有研究利用刺激抗her2免疫应答来消除her2阳性bc中的癌细胞。由于her-2/neu在许多正常细胞中以低水平表达,但在多种癌症中过表达,包括乳癌、卵巢癌、子宫内膜癌、胃癌、胰腺癌、前列腺癌和唾液腺癌,由于其过表达与肿瘤进展增强有关,以及对传统化疗的反应较弱,预后较差,该受体作为肿瘤相关抗原(tumor-associated antigen,taa)已成为癌症治疗的重要靶点。外显子19中的egfr基因缺失(del l747-s752)和密码子858(外显子21)中的常见点突变(l858r)已在非小细胞肺癌和腺癌中得到鉴定。5%的人肺鳞状细胞
癌具有egfrviii突变以及10-30%的肺腺癌具有egfr激酶结构域突变;研究显示,t790m、c797s突变及不同构型与肿瘤患者的耐药性、疗效预测、预后极其相关。egfr是在肺癌等多种实体瘤细胞中表达或异常表达、而在普通细胞中表达很微弱的细胞表面抗原;egfr与肿瘤细胞的增殖、肿瘤侵袭、转移及细胞凋亡的抑制有关;在癌症中,变异的egfr得到持续性的激活,从而引起细胞无节制的扩增并最终导致癌症。因此,egfr是一个癌症特异性靶点。研究表明,egfr-tki(酪氨酸激酶抑制剂)联合治疗在egfr突变模型中可促进t细胞浸润,降低肿瘤浸润treg和m2样巨噬细胞比例,提高对α-pd-1/pd-l1的反应性;其他联合策略包括α-pd-1/pd-l1 amg510(ras抑制剂)等。
[0085]
(2)b7-h3是来自b7家族的新型免疫检查点。b7同源蛋白(b7-h3,又称cd276)是b7家族新发现的免疫调节蛋白成员,由于其在肿瘤组织中过表达而在正常组织中表达有限,并参与肿瘤微环境(tumor microenvironment,tme)的形成和发展,成为具有吸引力和前景的肿瘤免疫治疗靶点。到目前为止,许多基于b7-h3的免疫治疗策略已经在临床前模型中证明了有效的抗肿瘤活性和可接受的安全性。靶向pd-1、ctla4等免疫疗法的成功,也为研究者开发以b7-h3为靶点的新免疫疗法提供了范例和方向。b7-h3在血清、癌前病变、肿瘤相关血管系统、csc、cic、肿瘤转移及复发中的作用,以及在诊断和治疗的中潜力,为未来基于b7-h3介导的肿瘤促进的精确细胞和分子机制探索,为肿瘤发展和免疫治疗的细胞生物学进步奠定基础。人类b7-h3基因已被定位到9号染色体,而小鼠b7-h3基因位于15号染色体上。人类b7-h3蛋白以跨膜或可溶性的亚型存在,跨膜b7-h3是i型跨膜蛋白,含有316个氨基酸,分子量约4566kda,由一个胞外结构域、一个跨膜结构域和一个短的胞内结构域组成;小鼠b7-h3(2igb7-h3,b7-h3 vc)的细胞外结构域由一对免疫球蛋白可变结构域和恒定结构域组成,而人类b7-h3(4igb7-h3,b7-h3vcvc)由于外显子复制而由两对组成。可溶性b7-h3(sb7-h3)被基质金属肽酶(mmp)从表面切割或通过内含子的选择性剪接产生,可在人类血清中检测到,在分泌体(如外泌体和其他细胞外囊泡)中也发现了b7-h3蛋白。trem-like transcript 2(tlt-2)已被鉴定为b7-h3的潜在受体,同时可能不是b7-h3的唯一受体。与其他免疫检查点相比,b7-h3不仅影响先天免疫和适应性免疫,而且还通过各种非免疫途径调节癌细胞的侵袭性。在大多数正常的人体组织中,b7-h3mrna广泛表达,而b7-h3蛋白相对较少,b7-h3 mrna和蛋白表达模式的差异表明,b7-h3具有严格的转录后调控机制。有证据表明,mir-124可能通过发挥肿瘤抑制作用并靶向b7-h3的3-utr引起翻译抑制;mir-29过表达可抑制b7-h3的表达水平,而b7-h3在促进成神经管细胞瘤血管生成中起着至关重要的作用;在结直肠癌(crc)中,mir-128负调控b7-h3的表达;同时,cd276在肿瘤细胞中mrna和蛋白水平均高表达。b7-h3增强侵袭性和迁移性的能力已经通过体外癌症模型进行了进一步研究,现有研究评估了b7检查点分子在卵巢癌(ovca)中的表达,发现b7-h3高表达,而不是pd-l1,并且b7-h3高表达与肿瘤浸润t细胞功能障碍相关。在t细胞激活过程中,b7-h3能有效且持续地抑制t细胞增殖和ifn-γ、il-13、il-10和il-2的产生。b7-h3除抑制t细胞外,还抑制nk细胞活性,转染4ig-b7-h3的cho-k细胞避免了nk细胞介导的细胞毒性。b7-h3形成的抑制性免疫微环境有助于癌症避免免疫破坏。除了在免疫途径中的作用,b7-h3还具有非免疫原性功能,如促进迁移和侵袭、抗凋亡、细胞活力、化疗耐药性和内皮-间充质(emt)过渡。在肿瘤细胞中,其还通过重要的细胞内信号转导通路参与重编程代谢。b7-h3也在葡萄糖代谢信号通路和重编程中发挥重要作用。癌细胞代谢的特点是糖酵解和乳酸生产的增加,即
使在丰富的氧气的情况下也是如此,这种现象称为warburg效应或有氧糖酵解。有氧糖酵解通过提供能量和生物合成材料,使癌细胞具有生长优势。研究表明,b7-h3通过ros介导的hif1a稳定调节糖代谢,以利于b7-h3增强肿瘤生长。有效消除b7-h3的异常表达特别是在癌症启动细胞(cics)上的表达,成为抗肿瘤治疗的必要条件。使用单克隆抗体阻断免疫检查点,ctla-4、程序性细胞死亡蛋白1(pd-1)和pd-1配体1(pd-l1)在各种癌症患者中显示出显著的临床成功。这种经验也可以应用到b7-h3。然而,由于缺乏人类b7-h3特异性阻断单克隆抗体,这一策略的临床应用受到了阻碍。基于b7-h3的肿瘤免疫治疗策略:(a)阻断单克隆抗体靶向b7-h3;(b)通过adcc靶向b7-h3;(c)通过adc治疗靶向b7-h3;(d)利用cd3结合的bsabs靶向b7-h3;(e)双特异性杀手接合器(bike)或三特异性杀手接合器(trikes);(f)小分子抑制剂靶向b7-h3;(g)car-t细胞和car-nk细胞靶向b7-h3;(h)与抗b7-h3疗法的协同选择。与抗b7-h3单抗化学偶联的抗cd3抗体已被临床批准;anti-cd3 anti-b7-h3双特异性抗体(b7-h3 bi-ab)被用于直接激活t细胞杀死肿瘤目标。双特异性杀手接合器(bike)或三特异性杀手接合器(trikes)在nk细胞和肿瘤细胞之间形成抗原特异性免疫突触,从而触发nk细胞介导的肿瘤细胞裂解。嵌合抗原受体(car)t细胞技术是靶向b7-h3进行免疫治疗的另一种有效方法,用car技术设计的自体t细胞或nk细胞,以肿瘤抗原为目标,并被过继地转移到患者体内以杀死癌细胞;在许多car-t治疗中,car-t细胞的活性依赖于抗原密度。最近,nk细胞被用来生成car-nk细胞,car-nk细胞可以控制移植到小鼠模型中的人类nsclc细胞的生长并延长生存时间。通过不同免疫疗法的联合治疗来提高患者的治疗效果和生存率越来越受到关注,多种免疫检查点抑制剂的联合疗法正迅速成为癌症治疗的一种手段。
[0086]
(3)新抗原tenm4具有重要作用。tenm4(teneurin 4)是由odz4基因编码的跨膜蛋白,位于11号染色体上,包含34个外显子,参与神经系统发育、神经突起生长和神经分化。tenm4突变和重排在诸多肿瘤中被发现,tenm4突变大部分发生在乳腺癌中,tenm4在细胞迁移和干细胞中发挥作用。caat增强子结合蛋白同源蛋白(chop)和糖皮质激素受体-α(grα)是实验建立的tenm4激活因子,但c-末端结合蛋白2(ctbp2)抑制其表达。其他与odz4启动子结合的蛋白包括myc和e2f转录因子1(e2f1),但在调节tenm4表达方面的作用尚未被证实。tenm4是一种糖基化ii型跨膜蛋白,属于teneurins(tenms)家族,其中包括4个tenm成员(tenm1-4);所有的tenm都具有很强的组织同源性,具有高度系统发育保守的c-末端胞外结构域,单通道跨膜序列,以及可变的小n端胞内结构域。与家族中的其他蛋白质一样,tenm4细胞外球状结构由几个结合基序组成,包括表皮生长因子(egf)类重复序列,它可以介导tenm4顺式二聚,然后是带有ncl-1的β螺旋桨区,ht2a和lin-41(nhl)结构域是同亲性tenm4反式作用所必需的,以及包含一个结合位点的天冬氨酸酪氨酸(yd)重复区是异亲性蛋白通信所必需的。与其他tenm一样,tenm4可以介导与g蛋白偶联受体家族latrophilins(lphns)以及肌动蛋白交联蛋白丝状蛋白的细胞相互作用。研究表明,与正常组织相比人肺腺癌、低级别胶质瘤和胰腺腺癌中tenm4mrna的表达显著增加,在肝癌、子宫内膜癌、胃癌、肾癌中tenm4的表达与预后呈相反的相关趋势,tenm4表达的升高与患者生存期的降低相关。tenm4蛋白在不同的人乳腺癌和小鼠乳腺癌细胞株中均有高表达,包括雌激素受体(er)、人表皮生长因子-2(her2)阳性乳腺癌和三阴性乳腺癌(tnbc)亚型(bt474、mcf-7、mda-mb-231、t47d、hcc-1806、4t1和zr7s)。此外,通过特异性sirna沉默tenm4与肿瘤球生成的显著受损和csc标志物(如cd44 /cd24细胞百分比)、八聚体结合转录因子4(oct4)、cd49f和醛脱氢酶
(aldh)活性的降低有关,说明在不同的乳腺癌亚型中,tenm4在肿瘤球形成和自我更新中发挥作用,并有助于乳腺癌细胞的恶性表型,可能通过促进转移进展和可能的耐药性。tenm4在除csc外的不同乳腺癌细胞亚型中过表达,并参与促进癌细胞迁移,提示tenm4可能与乳腺癌临床相关。在所有分析的乳腺癌亚型中,包括tnbc、her2阳性、孕激素受体(pr)阳性和er阳性,tenm4的高表达与较差的患者无复发生存(rfs)显著相关。tnbc中tenm4过表达与患者总生存期(os)较差显著相关,数据表明tenm4可能作为患者生存的生物标志物和可能靶点。由于tenm4是一种跨膜蛋白,在发育过程中表达受到限制,且在少数成人组织中发现低表达,因此被认为是药物和免疫治疗的良好靶点;其在分化的上皮细胞和未分化的csc中的表达意味着它的靶向可能有利于限制肿瘤负荷,但最重要的是其可以消除csc,从而限制肿瘤细胞的传播。编码tenm4的疫苗的开发将促进持久的体液免疫和细胞反应的产生,从而抵消乳腺肿瘤的进展。尿液中的tenm4检测是一种新颖的、信息量更大的方法,可以跟踪乳腺癌细胞释放tenm4,从而有助于建立一个新的乳腺癌检测、预后和治疗反应的生物标志物。mir-708可能是乳腺癌治疗的新靶点,也是乳腺癌患者治疗反应的一个预测性生物标志物;mir-708从tenm4mrna中剪接出来,加工并输出到细胞质中,加载到rna诱导的沉默复合体(risc)。
[0087]
(4)tenm4在乳腺癌发展和治疗中具有重要价值。现阶段乳腺癌的治疗是基于er、pr或her2受体的表达,靶向治疗在治疗某些乳腺癌亚型方面取得了相当大的成功。然而,tnbc是最具侵袭性的亚型之一,目前仍没有真正有效的靶向治疗方法。虽然原发性乳腺癌是高度可治疗的(80%-99%的被诊断为i/ii期乳腺癌的女性可存活到5年),目前还没有有效的治疗转移性乳腺癌的方法。基于此,本发明重点针对tnbc中新的潜在靶点的识别,并在该亚型中发现tenm4为一种新的肿瘤相关抗原。研究显示,tenm4除在小鼠和人的上皮tnbc肿瘤细胞上表达外,还在小鼠和人的tnbc-csc中进一步上调,且tenm4的表达并不局限于tnbc细胞,在其他乳腺癌亚型的细胞中也有表达。因此,在所有分析的乳腺癌亚型中,odz4在基因重排、突变和过表达方面均发生了改变,至少在tnbc-、her2-和pr阳性的患者中,odz4与较差的患者rfs和os存在临床相关性;tenm4过表达与3级乳腺癌患者更差的rfs和os相关,提示其可能与乳腺癌的侵袭性有关,可能作为一种新的潜在的预后和诊断指标具有临床作用。治疗维度显示,tenm4具有良好的肿瘤相关抗原特点,可以通过免疫方法进行靶向,包括抗癌疫苗;其免疫靶向不仅可以清除以上皮细胞为代表的大部分肿瘤,还可以清除肿瘤干细胞,这是乳腺癌转移和化疗耐药性的潜在原因。此外,tenm4作为一种跨膜蛋白,可被抗tenm4疫苗接种所诱导的体液和细胞反应靶向;tenm4通过抗癌疫苗接种,其低表达限制在神经系统,可以保证其安全性。因此,本发明首选tenm4作为乳腺癌的预防和治疗性疫苗,包括但不限于乳腺癌的预防和治疗。作为研究tenm4参与tnbc和her2阳性乳腺癌的起始和进展的有吸引力的模型,本发明已经证明了4t1、tubo和balb-neut小鼠模型肿瘤与tenm4的潜在相关性,以及其免疫靶向的临床影响。尽管它们在免疫学研究上的相关性有限,但将人乳腺癌细胞(如mda-mb-231)异种移植,可能对深入了解tenm4在人tnbc生物学、进展和转移中的作用至关重要。此外,tenm4免疫靶向对人类tnbc进展的影响也可以通过过继转移实验来阐明,该实验将免疫能力强的小鼠诱导的tenm4抗体和特异性t细胞注射到mda-md-231荷瘤小鼠体内。另外,考虑到mir-708在乳腺癌进展和治疗耐药性中的作用,进一步的研究评估该mirna在tnbc和her2阳性肿瘤的小鼠和人类临床前小鼠模型中的表达可能是有用
的。
[0088]
其中,car-t细胞治疗及car分子优化相关内容主要包括:
[0089]
嵌合抗原受体(car)-t细胞治疗在过去的十年中已经发展成为最有前途的癌症治疗方法之一,特别是针对血液肿瘤。car-t治疗包括使用car分子对患者的t细胞进行体外基因工程,使t细胞具有针对目标肿瘤细胞的定向特异性,并随后将car-t细胞注入患者体内进行癌症治疗。第一代car,通常包含用于抗原识别的细胞外单链可变片段(scfv),用于信号转导的铰链(h)和跨膜(tm)域,以及用于激活的细胞内cd3z。尽管存在抗原特异性激活和细胞毒性,第一代car-t细胞在体内显示低增殖。第二代car-t细胞在跨膜和cd3z结构域之间添加了共刺激结构域(例如cd28),并被证明可以解决第一代car-t细胞相关的低增殖问题。第二代car-t细胞治疗慢性淋巴细胞白血病(cll)和b细胞急性淋巴细胞白血病(b-all)患者的临床试验取得了前所未有的结果,包括完全缓解。第三代car,其特征是跨膜和cd3z结构域之间合并两个共刺激结构域(如cd28和ox40),也被开发并显示进一步增强car-t细胞的性能。然而,大多数car-t治疗的成功是在血液癌中。当面对实体肿瘤时,car-t细胞的治疗效果有限。主要是由于在免疫抑制肿瘤微环境(tme)中难以归位、浸润和生存,以及肿瘤抗原逃逸和异质性。此外,car-t治疗相关的副作用也有报道,包括靶向非肿瘤毒性、神经毒性、细胞因子释放综合征(crs)等。car-t细胞工程目标是(a)提高car-t细胞的疗效,以克服关于car-t治疗在实体肿瘤中的无效问题;(b)提高car-t细胞的安全性,以减轻和/或最小化与以前的car-t产品相关的不良毒性。为此,多种修改和优化策略已被开发并应用于组成表达的car。设计car分子包括:(a)car分子的外结构域、铰链结构域(h)、跨膜结构域(tm)和内结构域的修饰。(b)双特异性杀手接合器(bites)设计,或三特异性杀手接合器(trikes)。(c)设计利用双特异性t细胞参与(bites)作为car-t和肿瘤细胞之间的适配器的car-t。(d)设计cars实现基于蛋白开关的逻辑门,以整合来自两个或更多肿瘤抗原的输入。含有两个串联配体结合域的双特异性car已被证明在car-t信号转导中有效地实现or逻辑门,其中只有两种不同类型的抗原能够激活car受体。bcma/cs1双特异性car被证明比共同表达单独bcma和cs1单特异性car的t细胞表现更好。因此,双特异性car对靶向癌细胞的亲和力增强,可以增强免疫治疗,并有助于避免异质性肿瘤可能发生的抗原逃逸。通过修饰非car分子来改造装甲car-t细胞:(a)car-t细胞面对实体肿瘤的免疫抑制tme;(b)旨在帮助car-t细胞对抗免疫抑制tme的代表性方法,包括细胞因子、趋化因子受体、显性负性受体、肝素酶和crispr介导的基因组编辑和t细胞文库筛选的表达。细胞因子分泌重组白细胞介素(il)-12已被临床应用于多种类型的实体肿瘤。相关研究也开发了能够组成分泌il-12的抗卵巢肿瘤car-t细胞,并观察到这些装甲car-t细胞在抑制小鼠卵巢腹膜癌的tme中增强了生存。il-15培养的car-t细胞可以保留低分化的干细胞记忆(tscm)表型,减少衰竭,增强抗原结合时的增殖。包括巨噬细胞和树突状细胞在内的多种细胞类型都可以产生il-15,il-15可以刺激cd8 t细胞和nk细胞增殖,增加肿瘤细胞毒性。然而,仍然需要一种高通量的方法来筛选最有力地提高细胞治疗性能的候选基因。tgf-βr2 41bb嵌合受体,已被证明可以增强t细胞的适应性,促进关键效应因子的表达,并改善体内实体肿瘤清除。刺激诱导的car-t细胞,由外部刺激控制on和off开关,如肿瘤抗原、小分子、光、超声波和其他环境线索,已经被用来激活功能性car表达(on-switch)或失活car表达t细胞(off-switch)。自动化的car-t细胞基于合成的notch受体(synnotch)系统的and-gate t细胞工程,可以精确控
制car-t细胞信号通路。刺激诱导型car-t细胞的代表性设计包括:(a)同步car-t细胞。(b)rapalog诱导的car-t细胞。(c)蓝光可控的car-t细胞与lintad系统。(d)fus-可控性热敏car-t细胞。另一种自动条件表达cars的方法是由肿瘤微环境相关因素(如缺氧)驱动的。使用定量表征的缺氧反应元件(hre)和一组核心启动子。除了在诱导型car中使用外,小分子也被应用于移植前的car-t细胞预处理,以提高治疗效果。he等人也基于一个环状排列lov2(cplov2)的光学二聚化系统设计了optocar,并在体内证明了optocar t细胞的光诱导抗肿瘤活性。超声波和/或热可控的car-t细胞,可以深入生物组织。除了其传统的成像诊断作用,超声已被用于调节细胞功能的治疗目的。自然杀伤细胞(nk)是在先天性免疫中起关键作用的淋巴细胞。外周血标本中约10%的单核细胞为nk细胞。nk细胞通常对主要组织相容性(mhc)分子提出的抗原不敏感。因此,nk细胞可以被改造成异体治疗细胞产品,避免了gvhd。初步测试表明,car-nk很少引起crs。这对开发现成的治疗产品特别有吸引力。事实上,car-nk细胞已经被开发为对抗肿瘤的有效工具。从人的诱导多能干细胞中提取的nk细胞被设计表达car,并对卵巢癌表现出强烈的抗肿瘤活性。通过敲除细胞因子诱导的含sh2蛋白的基因,进一步工程这些ipsc衍生的nk-car细胞,可以显著增强nk-car细胞的代谢适应性,从而对多种类型的癌症具有更有效的毒性。car-nk具有毒性小、显著降低gvhd风险的优点,是一种更安全、更普遍的治疗方法。它的非抗原杀伤能力也可以用来解决某些肿瘤中抗原逃逸的问题。然而,car-nk在基因传递和细胞扩张方面也有困难。car-m和car-nk作为car-t治疗的替代和补充治疗还需要更多的优化。
[0090]
其中,联合治疗相关内容主要包括:
[0091]
针对程序性细胞死亡蛋白-1(programmed cell death protein-1,pd-1)或其配体pd-l1的抗体可以拯救t细胞,使其摆脱疲劳状态,恢复对癌细胞的免疫应答。现有联合疗法已经证实,包括α-pd-1/pd-l1加化疗、放疗、血管生成抑制剂、靶向治疗、其他免疫检查点抑制剂、共刺激分子激动剂、干扰素基因刺激剂激动剂、粪便微生物群移植、表观遗传调节剂、或代谢调节剂,具有优越的抗肿瘤功效和更高的反应率。此外,含有α-pd-1/pd-l1部分的双功能或双特异性抗体也具有更强的抗肿瘤活性。这些组合策略同时促进癌症免疫周期的多个过程,消除免疫抑制的缺陷,并协调免疫支持的肿瘤微环境。pd-1信号通常被癌细胞劫持以逃避免疫监视。当pd-1和t细胞受体(tcr)与其配体结合时,pd-1的免疫受体酪氨酸基抑制基序和免疫受体酪氨酸基开关基序被磷酸化。随后,src同源区域2domain-containing phosphatase(shp-2)被招募和激活,逆转tcr和cd28下游信号的磷酸化。pd-1除了抑制t细胞的一些早期激活途径外,还通过破坏tcr-pmhc-cd8的三分子相互作用直接破坏抗原识别。因此,pd-1信号通路抑制t细胞的功能,包括激活、增殖和细胞因子的产生。研究表明,egfr-tki在egfr突变模型中可促进t细胞浸润,降低肿瘤浸润treg和m2样巨噬细胞比例,提高对α-pd-1/pd-l1的反应性。此外,激活的egfr信号通路有助于癌细胞上pd-l1的上调,egfr-tki可能与α-pd-1/pd-l1协同作用减弱免疫逃逸。因此,egfr-tki α-pd-1/pd-l1治疗将使egfr突变癌症患者的免疫治疗疗效最大化。ras家族(kras,nras和hras)在癌细胞中经常发生突变。突变的kras是nsclc、结直肠癌和胰腺癌的一个确定的驱动基因。在正常细胞中,ras是由生长因子受体(如egfr)激活的。ras是一种小g蛋白,在gtp结合态(激活态)和gdp结合态(不激活态)之间切换。在激活状态下,ras会触发包括mapk和pi3k-akt在内的多个下游通路。在肿瘤细胞中,ras基因的突变会干扰gtp结合状态和gdp结合状
态之间的转换。因此,ras被锁定在gtp-bound状态,导致下游通路异常活跃,导致肿瘤生长。现有研究表明,ras及其下游途径参与了癌症免疫逃逸:负向调节癌细胞mhc-i的表达,增加细胞内在pd-l1水平,提高免疫抑制相关细胞因子的产生。ras靶向治疗可消除ras-mapk/pi3k-akt参与的免疫逃避,并与α-pd-1/pd-l1协同。lenvatinib(vegfr/fgfr抑制剂)联合α-pd-1在小鼠hcc模型中也显示出协同抗肿瘤作用。探索fgfr抑制剂联合α-pd-1疗效的临床研究仍在进行中。c-met也被称为肝细胞生长因子受体(hgfr)。激活的c-met信号会触发下游的mapk、pi3k-akt、rac1和fak通路。由于met突变、扩增或重排,c-met信号在多种癌症中过度激活。c-met信号可上调pd-l1的表达,而c-met抑制剂可使固有和ifn-γ诱导的pd-l1表达受损。shp2作为mapk、pi3k-akt、jak-stat和pd-1通路的汇聚节点,广泛调控细胞存活、免疫逃逸等多种癌症相关过程。抑制shp2可通过增强癌细胞内禀ifn-γ增加pd-l1和mhc-i的表达。shp2抑制剂可增强小鼠肿瘤模型中α-pd-1的疗效。现有shp2抑制剂联合α-pd-1的临床研究正在进行。鉴于cgas-sting通路在连接先天免疫和适应性免疫中的关键作用,sting激动剂如diabzi和msa-2已经被开发出来,可以系统给药。此外,锰还被鉴定为天然sting激动剂,在抗肿瘤免疫中发挥重要作用。sting激动剂 α-pd-1/pd-l1联合治疗可同时增强先天免疫和适应性免疫,有效克服免疫治疗耐药性。一方面,sting激动剂促进免疫细胞浸润,增强apc、nk和t细胞的功能。另一方面,α-pd-1/pd-l1抗体利用sting激动剂诱导的pd-l1上调。截至目前,sting激动剂联合α-pd-1/pd-l1的多项临床试验正在进行。双特异性/双功能抗体靶向pd-1/pd-l1已经成为联合治疗的一种选择。双特异性/双功能抗体同时用一种药物阻断两个分子。tgf-βpd-l1双特异性抗体ym101在免疫排斥肿瘤模型中也表现出强劲的抗肿瘤活性。目前已开发出大量双特异性抗体,大多数双特异性抗体在小鼠肿瘤模型中具有良好的抗肿瘤效果。此外,针对egfr 肿瘤的egfr pd-l1双特异性抗体,避免了在非恶性细胞上靶向/非肿瘤结合pd-l1。该抗体具有增强的肿瘤特异性,降低了抗肿瘤t细胞不加选择地重新激活和严重治疗相关不良事件的风险。这些肿瘤相关抗原pd-1/pd-l1双特异性抗体在疗效和安全性方面可能具有很大的优势。此外,pd-1pd-l1(ly3434172)和cd47pd-l1(ibi322)双特异性抗体相对于单特异性pd-1和pd-l1抗体具有增强的免疫调节性能和提高的抗肿瘤活性。嵌合抗原受体-t(car-t)细胞治疗 α-pd-1/pd-l1,可以以mhc独立的方式识别和结合癌症抗原。car-t细胞治疗提供了大量的癌症反应性t细胞,并克服了mhc下调介导的癌症免疫逃避。α-pd-1/pd-l1通过挽救car-t细胞衰竭来增强car-t细胞治疗。1期研究结果表明,car-t细胞治疗联合α-pd-1/pd-l1在恶性胸膜疾病患者中具有抗肿瘤活性。此外,可以分泌pd-1阻断单链可变片段(scfv)的car-t细胞通过自分泌和旁分泌的方式提高了抗肿瘤活性。这种联合策略保护car-t细胞免受免疫衰竭,并优化了car-t细胞的功效。因此,如何选择一个最佳的临床前模型是一个巨大的挑战,以确定联合方案的活性。相对于广泛应用的同基因小鼠模型,人性化患者来源模型可以提供更宝贵的疗效评价。本发明认为,应根据患者的免疫图谱和其他预测性生物标志物提供个体化联合治疗。一个整合基因组、转录组、免疫图谱、微生物组的综合框架可以用来选择从联合中受益的患者。对于非炎症性肿瘤患者,α-pd-1/pd-l1单药治疗难以提供临床益处,需要个体化联合治疗才能克服耐药性。在免疫排斥的背景下,tgf-β阻断剂等治疗可通过抑制caf活性和减少肿瘤周围胶原沉积来挽救受到抑制的t细胞穿透。在免疫缺失的背景下,放疗、化疗和sting激动剂等疗法可以通过诱导免疫原性细胞死亡、增加肿瘤抗原释放和促进apc功能来克服低
免疫原性介导的免疫耐受。这些疗法与α-pd-1/pd-l1结合,同时促进了癌症免疫周期中的多个过程,重塑了tme,大大促进了肿瘤从非炎症向炎症的转化。此外,随着双功能或双特异性抗体等下一代α-pd-1/pd-l1药物的开发,将大大延长α-pd-1/pd-l1治疗的适应症。
[0092]
本发明提出了一种基于mrna的肿瘤疫苗,该疫苗包括预防、治疗、免疫检查点疫苗和car-t细胞及免疫佐剂的mrna表达体及其脂质递送系统和联合抗癌方法。该mrna表达体以嵌合抗原的形式表达肿瘤特异性抗原、突变抗原、新抗原及car-t细胞受体和免疫佐剂。所述特异性抗原包括her2/neu,her2/neu和sars-cov-2野生或变体rbd抗原,新抗原tenm4,免疫检查点b7-h3,双特异性b7-h3嵌合抗原受体car分子及免疫佐剂il-15;所述脂质递送系统包括脂质体和脂质纳米颗粒系统;所述联合治疗方法,包括预防、治疗疫苗、免疫检查点疫苗或抗体、car-t细胞和佐剂的合理适当配伍治疗,达到全面全方位多基因多因素的精准肿瘤免疫治疗效果。
[0093]
实施例1
[0094]
本实施例为b7-h3/cd3 bite和b7-h3 car-t的构建及表达活性检测,通过使用共培养试验评估b7-h3/cd3 bite和b7-h3 car-t细胞的活性。
[0095]
(1)方法
[0096]
免疫荧光染色分析:细胞孵化24-well盘子盖玻片在37℃24h。标本沾60分钟4℃的主要抗体,然后沾fitc-conjugated二级抗体(美国proteintech rosemont,il)和dapi(beyotime生物技术研究所、上海、中国)。用于染色的主要单克隆抗体包括b7-h3(cell signaling technology,cst,danvers,ma,usa;d9m2l),cd70(abcam,cambridge,ma,usa;ab175389),tim-3(abcam;ab47997),vista(cst;d5l5t)、b7-h3单抗(j42)和j42-scfv-fc。图像是用共聚焦显微镜捕捉的。对于rna-seq和生存分析,数据来源于基因表达谱交互分析(ge-pia),且可访问癌症基因组图谱(tcga)。
[0097]
流式细胞术:检测b7-h3、cd70、tim-3、vista、icam-1、pd-1细胞表面表达水平。简单地说,细胞在冰冷的磷酸盐缓冲盐水(pbs)中清洗,然后离心收集。将细胞与人b7-h3、cd70、tim-3、pd-1、icam-1特异性单克隆抗体(biolegend,san diego,ca,usa;分别为dcn.70、113 16、f38-2e2、eh12.2h7和ha58)和vista(bd biosciences,franklin lakes,nj,usa;mih65)在黑暗的冰上放置30分钟。用冰冷的流式细胞仪缓冲液洗涤两次后,将细胞重悬于300μl的相同缓冲液中,并根据制造商的协议使用foressa流式细胞仪(bd biosciences)进行分析。抗人cd4和cd8单抗(bd biosciences;rpa-t4和rpa-t8)、cd3、cd45ro、ccr7、cd25、cd69、tim-3和pd-1(biolegend;hit3a,uchl1,g043h7,3c7,fn50,f38-2e2,eh12.2h7)用于t细胞表型和共培养分析。对于每个样本,至少有20,000个事件被采集并使用flowjo软件进行分析(v.10.6.0;https://www.flowjo.com)。
[0098]
(2)结果分析
[0099]
如图2所示,本发明将单克隆抗体mab-f22衍生的b7-h3单链抗体与cd3单链抗体结合,b7-h3/cd3 bite及b7-h3重定向的car-t细胞的构建(如图2a和图2b)。b7-h3/cd3 bite每个单链抗体包含相应的轻链(vl)和重链(vh),它们通过5-氨基酸(g4s)连接在一起(见图2a)。b7-h3重定向的car-t细胞的构建如图2b所示,增加了细胞内部分的共刺激活性。
[0100]
如图3所示,b7-h3单抗及其衍生的b7-h3-scfv-fc与snk-6细胞的结合特性见图3a,被免疫荧光所证实。图3b显示了纯化后mab和b7-h3 bite的sds page分析。通过流式细
胞术检测共表达的mcherry红色荧光蛋白(图3c中pe texas-red),证实了car-t细胞的转导效率。
[0101]
结果表明,与nt细胞相比,car的表达率约为60%。转导十天后的表型分析,b7-h3 car-t细胞用流式细胞仪显示表达率cd8 t细胞为58-62%和cd4 t细胞28-32%,(cd4 /cd8 t细胞的比例约为1:2),cd45ro没有显著差异,cd62l、cd69、cd25,pd-1或tim-3car-t细胞和nt细胞组之间无差别如(如图3d)。
[0102]
说明本发明生产的b7-h3 mab和scfv单链抗体,以及本发明构建的b7-h3 car-t细胞和b7-h3/cd3 bite,无论表达还是细胞的结合活力均很好。
[0103]
实施例2
[0104]
本实施例为b7-h3/cd3 bite和重定向car-t细胞体外诱导的t细胞介导细胞毒性检测,以进一步检验b7-h3/cd3 bite和car-t细胞的治疗有效性。
[0105]
(1)方法
[0106]
本发明通过细胞毒性作用检测。b7-h3重定向car-t细胞和b7-h3/cd3 bitemab的细胞毒性使用51cr分析。为了评价b7-h3重定向car-t细胞的作用,本发明通过铬酸钠(na251cro4)标记的ct-26细胞(1
×
105/ml)与效应细胞(car-t细胞和vehicle处理的对照t细胞)以效应靶比(e/t)为16:1、8:1、4:1和1:1的比例孵卵4小时。此外,b7-h3阴性raji细胞与效应细胞(car-t细胞和载体处理的对照t细胞),以上述不同的e/t比例孵育4小时。对于b7-h3/cd3bite,将1
×
105个细胞/ml的肿瘤细胞(snk-6和raji)与不同浓度的系列纯化b7-h3/cd3 bite或37℃的pbs预孵育1小时。将来自健康供体的pbmc以不同的e/t比值加入,37℃,5%co2孵育4小时。用伽马计数器测量上清液的放射性。按(试验释放自发释放)/(最大释放自发释放)为100。
[0107]
(2)结果分析
[0108]
在cr51释放细胞毒试验中,b7-h3/cd3 bite和b7-h3 car-t细胞在不同e/t比值下的特异性抗肿瘤作用结果如图4a和图4b所示。b7-h3/cd3 bite在ct26细胞中观察到剂量依赖性的特异性裂解,b7-h3/cd3 bite对ct26细胞的ic50为87.76ng/ml。b7-h3阴性raji细胞,即使有t细胞存在,如图4c所示,也未观察到细胞毒性。此外,载体转染的对照t细胞不介导杀伤。共培养后,用流式细胞仪对残余细胞进行流式细胞分析,结果如图4d,图4e所示。肿瘤残余细胞统计数据见图4d,细胞因子分泌见图4e。
[0109]
如图4所示,b7-h3 car-t细胞和b7-h3/cd3 bite可以显著降低ct26细胞的活力,但bite和car-t治疗组之间没有显著差异。此外,与载体转染的对照t细胞相比,b7-h3重定向的t细胞分泌ifn-γ、il-2增加14倍,tnf-α增加约20倍。同样,与pbs处理的对照组细胞相比,b7-h3/cd3 bite细胞的细胞因子水平显著增加(图4e)。这些数据表明,b7-h3 car-t细胞和b7-h3/cd3 bite的细胞溶解活性伴随着与t细胞诱导的细胞毒性一致的细胞因子释放。
[0110]
本结果进一步说明,本发明的b7-h3/cd3 bite和car-t细胞具有良好的t细胞介导的细胞毒性作用,也就是治疗活性和有效性。
[0111]
实施例3
[0112]
本实施例为b7-h3/cd3 bite和b7-h3重定向car-t细胞分别及联合在体内对模型动物抗肿瘤杀伤效果,以明确未来临床应用的有效性。
2可能与人类长期共存,因此,构建covid-19与肿瘤疫苗同时接种预防的联合疫苗,临床意义很大。
[0127]
另外,构建了ph7-b3的免疫检查点疫苗。h7-b3既构成免疫检查点,也同时在许多肿瘤高表达,而在正常组织表达很低。所以,本发明在构建h7-b3的bite和car-t细胞的同时,也构建了它的mrna疫苗,可以作为预防或治疗疫苗。
[0128]
本发明还构建了ptenm4新抗原mrna疫苗,该抗原同样在许多肿瘤中高表达,在正常组织中不表达或低表达。特别是在乳腺癌包括tnrc中高表达,为乳腺癌预防和治疗的联合疫苗提供研究基础。
[0129]
本发明还构建了一些细胞因子mrna表达载体,为癌症治疗疫苗的辅助手段使用提供研究基础,包括ifn-gamma,il-15等。
[0130]
上述所有载体均为慢病毒载体,所含mrna线性化后,纯化消毒和脂质体包裹形成mrna癌症疫苗。
[0131]
2)体内免疫原性检测:
[0132]
通过体外ifn-γ酶联免疫吸附试验,可检测单次注射上述疫苗诱导的小鼠有效t细胞免疫反应,来确定疫苗的有效性。用上述疫苗5
[0133]
×
108vp免疫小鼠(n=6,12只/组)3周后,用ifn-γ酶联免疫酶(elispot)检测小鼠脾细胞的t细胞应答,并通过聚合肽刺激细胞内细胞因子染色(ics)和荧光激活细胞分选(facs)分析,测定cd4 和cd8 t细胞反应。对单个疫苗的nags肽免疫原性反应显示为红色。多肽稀释剂dmso和刀豆球蛋白a分别作为阴性对照和阳性对照。三个独立实验的数据,用ifn-γics评估诱导t细胞应答的质量(cd4,蓝色圆圈,cd8,红色方块)(n=6只小鼠/组,代表三个实验)。
[0134]
(2)结果分析
[0135]
本发明构建的五个mrna癌症疫苗接种3周后t细胞活化百分比如图6所示。从图中结果可以看出,所有疫苗均有强烈的t细胞免疫反应,cd8 t细胞的活化百分比高于cd4 t细胞,符合预期。另外,her2/neu-mpv多表位疫苗,cd8 t细胞ifn-γ活化百分比明显高于其它疫苗,说明结合常见突变的多表位肽抗原,可能更有利于提高免疫原性和t细胞免疫反应。还有,her2/neu-sars-rbd疫苗,cd8 t细胞反应也明显增高,为预防肿瘤和sars-cov-2的联合疫苗提供研究基础。
[0136]
实施例5
[0137]
本实施例为早期接种预防性疫苗,预防肿瘤生长的检测,以检测接种预防疫苗后对肿瘤的预防作用。
[0138]
(1)方法
[0139]
为了确定基于mrna疫苗在体内的预防有效性,本发明进行了早期预防接种,疫苗通过肌肉注射给药,在股四头肌,以5
×
108vp,每侧50μl的体积。接种后两周用ct26肿瘤细胞攻毒,以评估疫苗的预防价值。
[0140]
注射到6-8周龄雌性scid小鼠的右侧腹皮下。将小鼠分为6组,60只小鼠(n=6
×
10/组)接种上述五种mrna癌症疫苗,免疫后2周,2
×
106ct26细胞注射到右下腹部,观察肿瘤生长情况。接种疫苗后28天,对处理和未处理(模拟mock)小鼠的肿瘤体积测量显示(双尾mann whitney u检验,p《0.0001)。
[0141]
(2)结果分析
[0142]
本发明的五种mrna癌症疫苗预防接种后两周进行ct26肿瘤细胞攻毒,接种疫苗后28天,接种组未见肿瘤生长(如图7),而模拟组10只小鼠,有9只长了肿瘤,肿瘤体积见图7中标注。说明所述五种mrna疫苗不但有t细胞免疫应答反应,更有良好的在体肿瘤预防作用。这种预防性干预在接种疫苗的小鼠中产生了100%的保护作用,而几乎所有未经预防的小鼠都出现了较大肿瘤。
[0143]
实施例6
[0144]
本实施例为转移肿瘤模型早中期治疗疫苗干预可控肿瘤生长(早中期疫苗治疗),以观察治疗性疫苗的早中期治疗效果。
[0145]
(1)方法
[0146]
①
、原发性肺转移瘤模型的早期疫苗干预
[0147]
制造早期原发性肺转移瘤模型,1
×
105个ct26细胞于接种前3天静脉注射至尾静脉。疫苗通过肌肉注射给药,在股四头肌,以5
×
108vp,每侧50μl的体积。第16天,肺灌注15%的印度墨水,收集并固定在fekete的溶液中。用解剖显微镜对肺表面的转移菌落进行计数。在已建立的定瘤实验中,即对未治疗组,分别注射2
×
106个ct26细胞或2
×
105个mc38细胞。疫苗治疗开始前(第0天),动物随机分组(肿瘤大小平均每组70-100mm3)。开始进行肺转移中期疫苗治疗干预。小鼠一旦出现痛苦迹象或肿瘤体积超过2000mm3就被杀死。每3-4天用数字卡尺测量肿瘤生长。肿瘤体积计算公式为:0.5length width2,其中length为较长的维度。
[0148]
②
、原发性肺转移瘤模型的中期疫苗干预
[0149]
如同上述早期干预模型组,作为对照的未处理组,即小鼠接种肿瘤后从第16天开始,小鼠按肿瘤体积随机分组(平均70-100mm3,n=6
×
10/组)。继续分为对照组和疫苗治疗组,处理当天作为第0天,如图8b,显示了肿瘤体积随时间的变化。
[0150]
③
、分析肿瘤小鼠的免疫反应。
[0151]
从未处理组及疫苗接种组分离til或脾细胞,并合并(n=4)。显示了多肽池再次刺激后测定的ifn-γ cd4 或cd8 t细胞的百分比。为了深入了解肿瘤耐药性的机制,本发明测量了大肿瘤小鼠的疫苗诱导的nags特异性t细胞应答。尽管在这种情况下缺乏有效性,疫苗诱导的nags特异性t细胞在接种后10天从肿瘤浸润中恢复,通过ics试验显示同源nas肽再次刺激后,在体外测量其效应功能。
[0152]
显示ifn-γ的有效产生(如图8c)的自发和疫苗诱导的t细胞都能浸润肿瘤,但不能控制已建立的肿瘤的生长。
[0153]
(2)结果分析
[0154]
通过制作肺转移肿瘤模型,观察了早中期疫苗干预治疗的效果,如图8所示。图8a显示早期干预(攻毒后第三天开始疫苗干预)的结果,与未干预组相比,五种疫苗干预组肿瘤的肺转移肿瘤结节数量明显少于对照组。其中her2/neu-pmv组,her2/neu-sars-rbd组和h7-b3组,效果更明显,p《0.01。
[0155]
中期干预组结果见图8b,在肿瘤接种攻毒后第16天开始疫苗干预,发现干预组与未干预组比较,肿瘤结节生长明显缓慢,上述三组的效果也更明显。说明即使在中期进行疫苗干预,对激发肿瘤免疫反应,延缓肿瘤生长都是有益的。特别发现,在中期干预第14天左
右,干预组肿瘤结节体积均有一个明显缩小过程(如图8b),说明干预后引起的免疫反应效果比较明显。
[0156]
中期疫苗诱导干预后,出现肿瘤体积的明显缩小,本发明对疫苗诱导的t细胞免疫反应进行了检测,发现通过ics试验显示同源nas肽再次刺激后,疫苗诱导的nags特异性t细胞在接种后10天恢复向肿瘤中浸润,在体外测量其效应显示,ifn-γ有效产生(如图8c)的自发和疫苗诱导的t细胞都能浸润肿瘤,成为肿瘤缩小的原因,但不能完全控制已建立的肿瘤生长。
[0157]
实施例7
[0158]
本实施例为对高负荷肿瘤的疫苗干预,即对晚期肿瘤的疫苗干预效果检测,由于晚期肿瘤的高负荷,所以需要结合pd-1进行治疗,这样也同时观察了联合治疗的效果。
[0159]
(1)方法
[0160]
高肿瘤负荷动物中的疗效需要与anti-pd1联合使用。本发明选择小鼠结肠癌ct26细胞系,因为它的新抗原(nag)负载高,对抗pd1治疗有反应。
[0161]
小鼠(n=12
×
10/组)接种ct26细胞。1周后,根据肿瘤体积随机分组,分别给予anti-pd1单独治疗和五种疫苗联合治疗。疫苗在第0天注射(i.m.),而anti-pd1则在第16天注射(i.p.),每周两次。在小鼠个体中,肿瘤随着时间的推移而生长。数据来自三个独立实验(双侧卡方检验;*p《0.05)。在疫苗和anti-pd1联合治疗的应答小鼠中,通过ifn-γelispot定量的na-特异性t细胞应答,分析对免疫原性新抗原(nag)的反应。用ifn-γics在疫苗和anti-pd1治疗的应答和非应答小鼠中测量nag特异性t细胞应答。在多肽池再次刺激后,第30天检测ifn-γ cd4 和cd8 t细胞的百分比显示。(双侧mann whitney u检验;**p《0.01n.s.,不显著)。
[0162]
(2)结果分析
[0163]
为了提高本发明新疫苗在肿瘤高负荷小鼠中的有效性,本发明评价了疫苗与anti-pd1联合治疗的效果。之所以选择anti-pd1,是因为它代表了最广泛使用的cpb,并被批准用于许多癌症适应症。anti-pd1和疫苗的联合治疗提供了显著的肿瘤控制,导致大约50%的小鼠肿瘤完全消退(如图9a和图9b),ct26建立的肿瘤对小部分治疗动物(15%)的抗pd1单药治疗有反应,显示肿瘤完全消退,非完全消退肿瘤的体积也明显缩小。另外,本发明仍然观察到,在使用pd-1治疗之前的单独疫苗治疗,也同样导致14天左右的肿瘤体积的明显缩小(如图9a和图9b),也说明了疫苗治疗的单独有效性。
[0164]
疫苗和抗pd1联合治疗的动物显示,在广度和效力方面,其诱导的系统性cd4 和cd8 t细胞反应均较强烈。应答组与非应答组小鼠的nags特异性t细胞反应比较,显示应答组中ifnγ cd8 t细胞水平显著升高(如图9c)。研究表明,cd8 t细胞的选择性缺失会完全消除机体的抗肿瘤作用,凸显了这一淋巴细胞群对治疗效果的贡献。本发明实施例结果表明,cd8 t细胞在肺转移的早中晚期治疗中均发挥了关键作用,本发明的mrna癌症疫苗,具有显著升高ifnγ cd8 t细胞水平的作用。
[0165]
实施例8
[0166]
本实施例为抗肿瘤的联合治疗效果检测,即对上述经过疫苗预防和治疗无效的肿瘤(无反应者),以及加用pd-1治疗仍然未治愈的肿瘤,采取本发明的所有疫苗的联合治疗,再加用pd-1、加用b7-h3car-t细胞和辅助刺激因子如il-15等全面联合治疗,达到多表位、
多抗原及多基因多因素的综合治疗,以观疗效。
[0167]
方法及结果分析:
[0168]
对单个疫苗联合pd-1治疗无效的小鼠肿瘤进行dna-外显子组测序和rnaseq,结果表明所有免疫原性突变仍然存在,并在无反应的小鼠中表达。这表明这些动物对治疗缺乏反应并不是因为nag缺失。对治疗的抵抗与mhc-i基因缺失或mhc表达缺失无关。基因表达谱也作了评估。在抗pd1组、单独疫苗组或联合疫苗组中,来自无应答者的肿瘤的转录谱与未经治疗的肿瘤转录谱没有区别。然而,当比较抗pd1组和单个疫苗联合组应答者与未治疗肿瘤的基因表达时,观察到有显著的差异表达基因(deg)存在。其中大多数与联合治疗组相同。
[0169]
本发明进一步发现,联合治疗确实导致了肿瘤内t细胞库的多样化,接种疫苗本身并没有导致肿瘤中这种不同的克隆型的富集,这表明在没有抗pd1治疗的情况下,疫苗诱导的t细胞缺乏扩增。进一步通过转录组来评估不同治疗组中tcr克隆型的数量变化。发现肿瘤内选定t细胞克隆的扩张与反应相关。联合治疗显著增加了特异性调节的基因数量。这可能与联合治疗的免疫反应和疗效增加有关。
[0170]
本实施例表明,联合治疗确实增加了对无反应肿瘤的治疗效果,如图10a所示,当所有疫苗混合治疗 pd-1,再联合b7-h3 car-t细胞治疗,效果明显好于联合il-15;若进一步将car-t细胞与il-15联合混合疫苗和pd-1治疗,实现了肿瘤的100%消退(其他组肿瘤体积缩小,但不能完全消退)。如图10b显示,联合治疗增加了动物体内的特异性克隆型的数量,这可能是免疫反应和疗效增加的基础。
[0171]
总之,本实施例说明,本发明的预防疫苗,也可以联合起来作为混合治疗疫苗使用,如果进一步联合本发明b7-h3 car-t细胞治疗,效果更佳。
[0172]
应当说明的是,上述实施例均可根据需要自由组合。以上介绍仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。