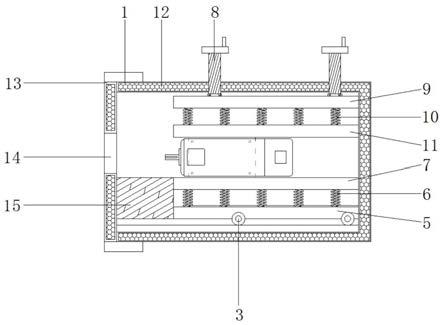
1.本技术涉及人参/红曲霉发酵物的制药用途,具体地,本技术涉及人参/红曲霉发酵物在制备用于调节和优化肠道菌群的药物中的用途。
背景技术:
2.肠道菌群是寄居于宿主肠道内且与宿主互利共生的微生物群体,根据系统发育地位分类,可将肠道菌群分为厚壁菌门、拟杆菌门、变形菌门、放线菌门、疣微菌门和梭杆菌门6个门。肠道菌群的组成受饮食、环境和疾病等多种因素的影响。随着dna测序技术的发展,众多研究发现,肠道菌群与人体健康存在密切关系。肠道菌群的改变可以作为某一特定疾病发生的标志。
3.研究证实肠道菌群、高脂饮食、hlp三者间存在紧密联系,肠道微生物在调控机体能量消耗和脂质代谢等方面发挥着非常重要的作用。其中ba分子是机体与肠道微生物相互作用的纽带。初级ba(主要是鹅去氧胆酸或胆酸)在肝脏中合成会进入肠道,在肠道细菌的去结合、差向异构或脱羟基作用下产生次生ba(主要是脱氧胆酸或石胆酸),并在回肠末端重新吸收到肝门静脉循环中。
4.ba可直接对肠道细菌的生长及菌群组成产生调节作用。胆管阻塞导致的胆汁淤积无法进入肠道,会引起肠道中细菌过度增殖,肠道黏膜屏障被破坏,病原菌内毒素入血,引起器官及组织的炎症和损伤。vicente等发现口服胆汁酸可显著抑制结肠溃疡模型大鼠肠道病原菌的过度增殖,从而降低血清内毒素的浓度。然而,过量的胆汁酸也会抑制肠道有益菌属的生长,破坏肠道菌群的构成。yokota等发现,高脂饮食会增加胆汁酸的排泄从而削弱肠道菌群整体数量和多样性的,升高厚壁菌门/拟杆菌门的比值。胆汁酸是高脂饮食改变宿主微生物菌群形态结构的决定性因素,只有适应能力强的种属才能保持并扩大自己的菌群优势。
5.最近,joyce等研究表明,肠道微生物影响机体能量和脂质代谢的关键在于细菌胆盐水解酶(bile salt hydrolase,bsh)。该酶由特定种属的肠道微生物产生,目前已知在乳酸菌属(lactobacillus)、双歧杆菌属(bifidobacterium)、肠球菌属(enterococcus)、梭菌属(clostridium spp)和拟杆菌属(bacteroides)等中均能产生bsh,bsh促进结合胆汁酸分子水解,产生次级胆汁酸,进而调节肠内fxr受体及其下游通路参与机体胆汁酸循环和糖脂代谢。
6.人参(panax ginseng c.a.meyer)为五加科植物人参的干燥根和根茎,最早载于我国的《神农本草经》,是我国传统的名贵中药。人参具有大补元气、复脉固脱、调补五脏、生津止渴、安神益智等功效,其主要化学成分为人参皂苷、多糖、黄酮等。其中人参皂苷在人参药用活性成分中占有重要地位,具有广泛的药理作用。
7.人参具有良好的降脂功效,中医认为人参的降血脂功效主要通过健脾益气发挥作用。朱丹溪将之述为“理脾如烈日当空,痰浊阴凝自散”。在许多经典方剂中,人参都发挥了重要作用。赵保胜等用人参白虎汤作用于糖尿病大鼠,研究发现血清中的tc、tg含量明显降
低,hdl-c含量升高,表现出有效的降血脂作用。陈湘君等从健脾化痰利湿角度出发,发现自创的人参降脂合剂可有效降低hlp患者的血脂水平。
8.红曲霉(monascus ruber)属于真菌界(fungi),子囊菌门(ascomycota),散囊菌亚纲(eurotiomycetes),曲霉科(aspergillaceae),红曲霉属(monascus)。它作为药食两用真菌在我国的应用已有千年的历史。由红曲霉发酵而来的中药红曲自古以来就被中国医家视为珍贵的中药和保健补品。中药红曲性温、味甘,具有活血化瘀、温中止痢、消食降脂等功效。
9.双向发酵技术是上世纪80年代庄毅教授提出的一种新型发酵手段,其技术的关键是以中药作为基质进行药用真菌的发酵生产,对中药而言具有扩大药性、增强药效、降低毒性等作用。研究表明,红曲霉在发酵过程中可产生大量的酶系,其中β-葡萄糖苷酶是一种纤维素水解酶,能够水解人参皂苷上的β-葡萄糖苷键,使其皂苷发生转化。丛悦怡等研究表明红曲霉与人参进行发酵可以提高总皂苷的质量分数,同时还可以使人参皂苷发生转化。厍守权发现,人参-红曲霉的固态发酵产物具有一定的降脂活性,并且对hela细胞的增殖有明显的抑制作用。但对其发酵产物的机制研究并未进一步阐明。因此,深入研究人参/红曲霉双向发酵体系药理作用机制具有一定的意义与价值。
技术实现要素:
10.本发明的技术目的是提供人参/红曲霉发酵物的新制药用途。
11.本发明提供了人参/红曲霉发酵物在制备用于调节和优化肠道菌群和/或用于治疗肠道菌群失调的药物中的用途。
12.在具体实施方式中,所述调节和优化肠道菌群包括使肠道有益菌属增加,有害菌种减少。
13.在具体实施方式中,所述人参/红曲霉发酵物通过调节肠道微生物多样性和丰度来调节和优化肠道菌群。
14.在具体实施方式中,所述调节和优化肠道菌群包括使普雷沃氏菌属的丰度增加,使muribaculaceae属的丰度降低。
15.在具体实施方式中,所述调节和优化肠道菌群包括降低firmicutes/bacteroidetes的比值。
16.在具体实施方式中,所述肠道菌群失调与高脂血症相关。
17.在具体实施方式中,所述肠道菌群失调由高脂血症引起。
18.另一方面,本发明提供一种人参/红曲霉发酵物在制备用于治疗与肠道菌群失调相关疾病的药物中的用途。
19.在具体实施方式中,上述人参/红曲霉发酵物是通过以下方法制备的:
20.(1)将红曲霉活化制成种子液;
21.(2)将干人参加水制成人参匀浆,经灭菌冷却后,将步骤(1)中得到的种子液接种到人参匀浆,发酵培养得到发酵液,将所述发酵液冷冻干燥即得发酵产物。
22.上述方法步骤(2)中,优选地,人参匀浆中人参添加量可为0.5-3wt%,优选2wt%。
23.上述方法步骤(2)中,优选地,种子液的接种量为2-5体积%。
24.有益效果
25.本技术提供了人参/红曲霉发酵物在用于调节和优化肠道菌群和/或用于治疗肠道菌群失调中的新用途。本技术通过实验验证了人参/红曲霉发酵物可以有效改善肠道菌群的结构,可能通过特定增加或降低某一菌属水平,例如使有益菌属增加,有害菌种减少,来实现其功效。
附图说明
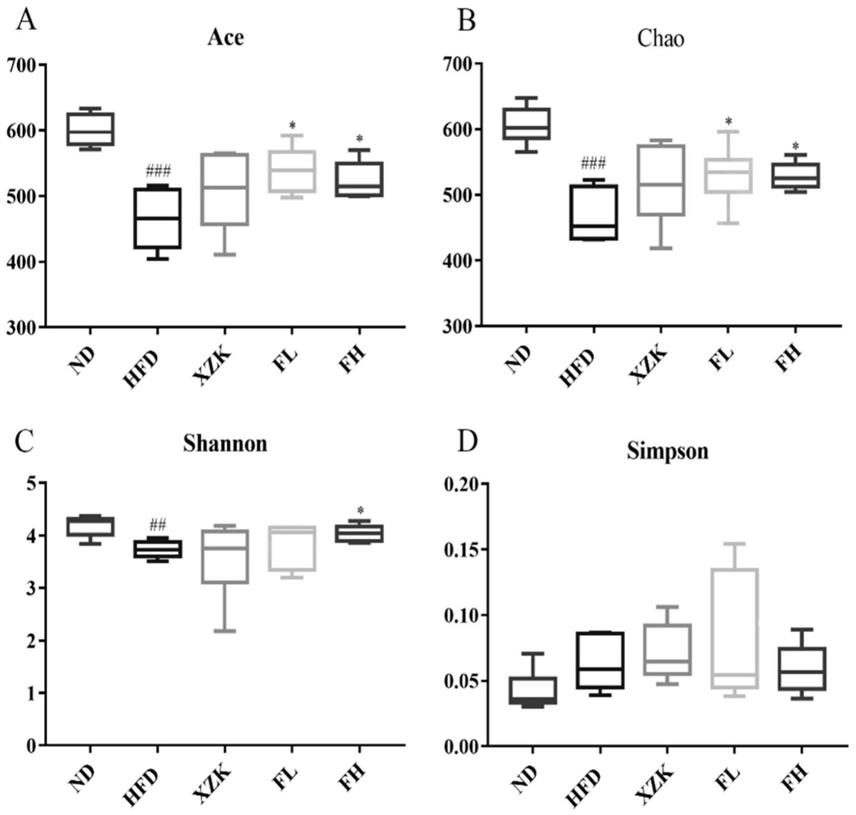
26.图1:alpha多样性指数(a.ace指数,b.chao指数,c.shannon指数,d.simpson指数)。数据以平均值
±
标准误差表示。#代表hfd组与hd组之间的差异(#:p<0.05,##:p<0.01,###:p<0.001),*代表各给药组与hfd组间的差异(*:p<0.05,**:p<0.01,***:p<0.001)。
27.图2:大鼠肠道微生物物种组成分析(a.物种在门水平上的相对丰度,b.firmicutes门/bacteroidetes门的比值,c.群落结构差异及聚类分析heat map图,d.otu venn图分析结果)。数据以平均值
±
标准误差表示。#代表hfd组与hd组之间的差异(#:p<0.05,##:p<0.01,###:p<0.001),*代表各给药组与hfd组间的差异(*:p<0.05,**:p<0.01,***:p<0.001)。
28.图3:pca主成分分析图,a:x轴和y轴表示两个选定的主坐标轴,百分比表示主坐标轴对样本组成差异的解释度值,不同深浅颜色或形状的点代表不同分组的样本,两样本点越接近,表明两样本物种组成越相似。b:x轴代表pc1轴上的差异离散情况,y轴表示不同样品组。
29.图4:anosim分析图。
30.图5:属水平组间差异显著性检验。注:纵轴表示属水平下的物种名,横轴表示该物种的平均相对丰度。最右侧为p值,*代表组间存在显著差异p《0.05;**代表p《0.01;***代表p《0.001。a:nd,hfd,xzk,fl,fh五组之间的差异。b:nd与hfd之间的差异。c:nd与fh之间的差异。d:hfd与fh之间的差异。
31.图6:lefse多级物种差异判别分析。a:进化分支图,不同深浅颜色节点表示在对应组别中显著富集,且对组间差异存在显著影响的微生物类群;深黑色点表示在不同分组中均无显著差异,或对组间差异无显著影响的微生物类群。b:lda判别条形图,统计各组中有显著作用的微生物类群,lda分值越大,代表物种丰度对差异效果影响越大。
具体实施方式
32.制备实施例
33.1.红曲霉的活化及种子液的制备
34.将保存在斜面中的红曲霉(monascus ruber,保藏编号为:cgmcc3.4450)接种于固体pda培养基(市售的马铃薯葡萄糖琼脂液体培养基)上,置于温度28℃,湿度80%的恒温恒湿箱中培养7天。挑取活化好的菌种接种于pda液体培养基中,置于温度28℃,转速160rpm的摇床上培养5天,制得种子液。
35.2液态发酵体系的建立
36.取1g干人参,洗净,加100ml水匀浆,灭菌。将种子液以5体积%接种量接种于人参匀浆中,置于温度为28℃,转速为160rpm的摇床上培养6天,待发酵完成后,将发酵液冷冻干
燥即得发酵产物,称量干重后密封保存,置于-20℃冰箱中备用。
37.实验实施例
38.i.实验部分
39.1.药物的制备
40.将上文制备的冷冻干燥的发酵产物粉碎成粉末状,得人参/红曲霉发酵产物(下文称为pm)。加去离子水制备成浓度分别为10mg
·
ml-1
和20mg
·
ml-1
的混悬液,现配现用。
41.2.动物的饲养及分组
42.雄性sd大鼠35只,购入后观察动物的体重、进食状况及一般生理指标,自由进食饮水,12h昼夜光线照明,室温26℃,相对湿度40%-60%。适应性喂养一周后随机分为正常对照组(nd,n=7)和模型对照组(n=28),正常对照组给予正常饮食喂养,模型对照组给予高脂饲料(63.6%基础饲料 15%猪油 20%蔗糖 1.2%胆固醇 0.2%胆酸钠)喂养,4周后测定各组大鼠血脂水平(与正常组相比有显著性差异表示模型构建成功)。
43.模型构建成功后,将模型对照组按体重平均随机分为高脂模型组(hfd,n=7)、血脂康对照组(xzk,n=7)、发酵物低剂量组(fl,n=7)和发酵物高剂量组(fh,n=7)。各组分别给予不同药物处理。根据血脂康胶囊成人口服用药剂量,以比表面积换算为大鼠用药剂量,高低剂量组给药浓度为2倍关系。xzk组给予血脂康胶囊200mg
·
kg-1
·
d-1
,fl组给予pm 200mg
·
kg-1
·
d-1
,fh组给予pm 400mg
·
kg-1
·
d-1
。nd组和hfd组均给予生理盐水灌胃。给药周期为4周,灌胃体积为2ml
·
100g-1
。隔天记录动物的体重及摄食量。
44.3.样品的收集及处理
45.无菌条件下收集各组大鼠粪便,根据粪便dna提取试剂盒的说明书进行大鼠粪便dna的提取,提取完成后,测定每个样本的dna浓度,利用1%琼脂糖凝胶电泳检测dna条带完整性,最后将合格的dna样本置于-20℃下保存或立即送往测序公司进行测序。
46.4.数据处理
47.由于测序所得到的原始数据(raw data)存在一定比例的干扰数据(dirty data),为了使菌群信息分析的结果更加准确、可靠,首先要对原始数据经过拼接、过滤、去嵌合体后,得到有效序列(effective tags)。将序列按照彼此的相似性分归为的最小分类单元otu(operational taxonomic unit),对97%相似水平下的otu进行聚类分析,并利用silva数据库进行物种注释。利用usearch(vsesion 7.0http://drive5.com/uparse/)软件平台进行otu聚类,采用rdp classifier贝叶斯算法对97%相似水平的otu代表序列进行分类学分析,并在各个水平domain(域),kingdom(界),phylum(门),class(纲),order(目),family(科),genus(属),species(种)上统计每个样品的群落组成研究微生物的多样性。otus聚类的结果可对每一个otu的代表序列进行物种注释,得到对应的物种信息和基于物种的丰富度分布情况。
48.在out水平下,单样本的多样性(alpha多样性)分析可以反映物种丰富度和均匀度等信息,常用的度量标准包括sobs、chao、ace、shannon、simpson、coverage指数。venn图分析可以得到不同样品或分组间的共有和特有的otus信息等。群落组成分析包括群落bar图、pie图、heatmap图等,可以得到不同分组在各分类水平上的物种组成情况及优势物种的相对丰度。upgma样品聚类树、pca、pcoa及nmds是根据不同的距离算法分析不同样本群落结构的相似性和差异,以上各个方法是对同一结果采用的不同的描述一般用r软件进行。
49.组间显著性差异检验是基于群落丰度数据,运用严格的统计学方法对不同组微生物群落之间的物种进行假设检验,用来评估物种丰度差异的显著性水平,以获得组间显著性差异物种。多组独立样本采用单因素方差分析(one-way anova),两组样本间比较采用student t检验(方差相等)或welch t检验(方差不等)。lefse评估微生物群落间的差异是否符合预期的生物学行为,通过检测有显著丰度差异特征,并找到与丰度有显著性差异的类群,采用线性判别分析(lda)来估算每个组分(物种)丰度对差异效果影响的大小。
50.ii.结果分析
51.1.otu聚类结果
52.本技术采用illumina mi seq测序平台对粪便菌群样本16s rrna基因的v3-v4区进行测序,共得到1,337,123条有效序列,每个样品平均含有43,133条有效序列。将有效序列按照相似性的不同分归为不同的out进行生物信息统计分析。
53.2.肠道微生物多样性分析
54.研究肠道微生物的多样性,可以采用单样本的多样性(alpha多样性)分析反映微生物群落的丰富度和多样性。coverage指数反映群落覆盖度(community coverage),该指数反映本次测序结果是否代表了样本中微生物的真实情况。结果显示,各组大鼠粪便dna测序结果中的coverage指数均在99%以上,说明各组大鼠的测序深度满足需求,几乎没有未被检测出的序列。
55.ace指数和chao指数分别通过估计群落和样本中所含otu数目反映群落丰度(community richness),其数值与群落丰度成正比。shannon指数和simpson指数可以用来反映群落微生物多样性(community diversity)。shannon值越大,说明群落物种种类越丰富,种类中个体分配上的平均性或均匀性也越高;而simpson指数值越大,则说明群落均匀度越低。观察各组大鼠alpha多样性各指数的箱型图发现(见图1),相比于正常组,hfd组ace指数和chao指数数值均显著下降(p《0.001),说明高脂饮食喂养导致大鼠肠道菌群物种丰度减少。给药治疗后,与hfd组相比,xzk组、fl组和fh组ace指数和chao指数均上升,其中fl组和fh组具有统计学差异(p《0.05)。这表明不同剂量的pm均可以有效增加hlp大鼠肠道菌群的物种丰度,且效果优于阳性对照药物。shannon指数和simpson指数见图1c和d,与nd组相比,hfd组shannon指数降低,simpson指数升高,说明hfd组物种多样性减少,均匀度降低。在给予药物治疗后,xzk组shannon指数无明显改变,fl组fh组均有不同程度的上升,且fh组有显著性差异(p《0.05)。除xzk组外,fl组和fh组simpson指数均下降,但无统计学差异(p》0.05)。表明高剂量pm治疗可以有效提高hlp大鼠肠道菌群物种的多样性及均匀度。
56.综上alpha多样性分析结果表明,pm可有效增加hlp大鼠肠道微生物群落丰度,并在一定程度上恢复大鼠肠道菌群的多样性及均匀度,而阳性对照药物对此无显著效果。
57.3.肠道微生物物种组成分析
58.群落bar图是根据分类学分析结果,得到不同分组在各分类水平(如域、界、门、纲、目、科、属、种、otu等)上的物种组成情况,可以直观展现各样本在某一分类学水平上含有哪些优势物种,以及样本中各优势物种的相对丰度。图2a为各组大鼠肠道菌群的序列在门水平上的优势物种及丰度情况。粪便菌群在门水平上主要被划分为厚壁菌门(firmicutes)、拟杆菌门(bacteroidetes)、变形菌门(proteobacteria)、epsilonbacteraeota门、放线菌门(actinobacteria)以及部分相对丰度较低的细菌。其中厚壁菌门和拟杆菌门为主要优势
菌门,丰度最高。研究表明,肥胖的发展可能与肠道菌群中厚壁菌门与拟杆菌门的比例增加,菌群的多样性减少有关。由图2a和b可知,高脂饮食会造成厚壁菌门丰度增加,拟杆菌门丰度降低,相比于nd组,hfd组厚壁菌门/拟杆菌门的比值显著升高(p《0.01),给予药物治疗后,xzk组无显著性改变,而fl组和fh组这一比值显著降低(p《0.05),这表明不同剂量pm治疗能够有效降低厚壁菌门丰度,同时增加拟杆菌门丰度,且效果优于阳性对照药物。
59.对属水平上的物种注释及丰度信息进行聚类,将丰度排名前50的菌群绘制成热图,以反映每组大鼠肠道菌群结构组成的相似性和差异性,结果见图2c。图中不同深浅颜色代表不同物种的丰度大小。由图可知,nd组在属水平上丰度排名前四的菌属为muribaculaceae属(0.14%)、lachnospiraceae_nk4a136属(0.12%)、prevotellaceae_nk3b31属(0.11%)和乳杆菌属(0.08%);hfd组与xzk组相同为巨单胞菌属(0.12%和0.27%)、muribaculaceae属(0.11%和0.09%)、乳杆菌属(均为0.08%)和罗斯氏菌属(0.08%和0.07%);fl组为巨单胞菌属(0.19%)、muribaculaceae属(0.13%)、bacterioides属(0.07%),blautia属(0.05%);fh组为巨单胞菌属(0.12%)、blautia属(0.12%)、乳杆菌属(0.11%)和普雷沃氏菌属(0.11%)。结果表明,各组样本丰度菌属均存在差异。基于群落组成对各组样本在属水平上进行层次聚类分析,结果显示,fl组和xzk组肠道菌群属水平上群落组成相似度最高。
60.venn图可用于统计多组或多个样本中所共有和独有的otu数目,比较直观地呈现环境样本的otu组成相似性及重叠情况。由图2d可知,nd组out总数为796个,其中独有otu数量为156个;hfd组out总数为673个,其中独有otu数量为3个,nd组与hfd组共有的otu数目是496个,这说明高脂饮食减少了肠道菌群的out数目,改变了out的组成。在给予药物干预后,xzk组独有otu数量为103个,fl组和fh组独有otu数量分别为11个和29个,xzk、fl组和fh组与nd组共有的otu数目分别增加到560个、548个和552个,均高于hfd组与nd组共有out数目(496个)。这表明各给药组肠道菌群的out组成与正常样本的相似度得到了提高。
61.4.pca主成分分析
62.主成分分析(pca,principal component analysis)是β多样性的常用分析方法。该法应用方差分解对多维数据进行降维,从而提取出数据中最主要的元素和结构。样本物种组成越相似,则在pca图中的距离越近。如图3a所示,在out水平上,各组组内样本在pca图中聚类较好,说明各样本组内物种组成相似度高,分组合理。
63.基于pca分析结果,将不同分组样本在第一主成分(pc1)轴上做箱线图(图3b),直观呈现不同分组样本在pc1轴上的差异离散情况。各组样本中位值越接近,表明样本物种组成较相近。与nd组相比,hfd组、xzk组和fl组在pc1维度上中位数值相距较远,说明物种相似度相差较大,fh组与nd组相距较近,其物种相似度较高。
64.5.物种差异分析
65.anosim分析用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。它利用距离矩阵对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对划分的统计学意义进行显著性分析。对各组样本进行属水平的anosim分析,根据图4可知,between组距离值显著大于其他五组物种距离值,得到r值为0.5019,p值为0.001(r值大于0,说明组间差异显著,p值小于0.05表示统计具有显著性)。说明组间差异显著大于组内,分组有意义。
66.进一步对5组样本组间差异性显著的优势属进行分析,多组比较结果显示(图5a):在属分类水平上,种群间差异显著的细菌为巨单胞菌属(megamonas)、muribaculaceae属、blautia属、罗斯氏菌属(roseburia)、毛螺旋菌科nk4a136组(lachnospiraceae_nk4a136 group)、普雷沃氏菌属_9(prevotella_9)、普雷沃氏菌科nk3b31组(prevotellaceae_nk3b31_group)、粪杆菌属(faecalibacterium)、普雷沃氏菌属_1(prevotella_1)、考拉杆菌属(phascolarctobacterium)、螺杆菌属、瘤胃球菌属(ruminococcus)、allobaculum属、普雷沃氏菌科ucg-001组(prevotellaceae_ucg-001group)、粪球菌属。对nd组、hfd组和fh组进行两两比较,nd组与hfd组之间有7种菌属间存在显著性差异。其中,hfd组的巨单胞菌属、罗斯氏菌属、普雷沃氏菌属_9、blautia属和考拉杆菌属水平显著高于nd组(p《0.05),而nd组的普雷沃氏菌科nk3b31组和瘤胃球菌属两种属水平显著高于hfd组(p《0.05);fh组的巨单胞菌属、blautia属和普雷沃氏菌属_9水平显著高于nd组(p《0.05),而nd组的muribaculaceae属、毛螺旋菌科nk4a136_组、普雷沃氏菌科nk3b31组和瘤胃球菌属这4种属水平显著高于fh组(p《0.05);在hfd组和fh组只有muribaculaceae属和普雷沃氏菌属_9有显著性差异,hfd组muribaculaceae属水平显著高于fh组(p《0.05),普雷沃氏菌属_9水平显著低于fh组。结果表明,高脂饮食喂养会导致肠道内微生物群菌属产生较大差异。高剂量pm治疗会引起muribaculaceae属和普雷沃氏菌属_9两种菌属丰度发生较大的变化。推测muribaculaceae属和普雷沃氏菌属_9可能是pm发挥治疗作用的关键菌属。
67.6.lefse多级物种差异判别分析
68.lefse是根据分类学组成对样本按照不同的分组条件进行线性判别分析(linear discriminant analysis,lda),找出对样本划分产生显著性差异影响的群落或物种。首先使用秩和检验检测具有显著丰度差异特征,并找到与丰度有显著性差异的类群。然后,采用lda来估算每个组分(物种)丰度对差异效果影响的大小。lefse的分析结果主要包括两方面:进化分支图和lda判别柱状图。
69.在进化分支图中(见图6a)中,由内至外的圈代表了由门至科的分类级别,每一个圆圈的直径大小与对应的物种的相对丰富度成正比。图中展示了各分组中重要的微生物类群。进化分支图中的物种与lda判别柱状图(图6b)成对应关系。lda判别柱状图展示了lda大于设定值(默认设置为2)的物种。图中可以观察到不同分组间有丰度差异的物种,柱状图的长度代表差异性物种的影响大小,结果显示nd组中包括梭状芽胞杆菌纲、拟杆菌门、瘤胃球菌科、普雷沃氏菌科、muribaculaceae科、巴斯德氏菌目、clostridiales目、柔膜菌门、理研菌科,hfd组中包括红蝽杆菌科、氨基酸球菌科、脱铁杆菌门、产氧光细菌纲、γ-变形菌纲、eggerthellaceae科,xzk组中包括negativicutes纲、selenomonadales目、韦荣氏菌科、厚壁菌门,fl组中包括丹毒丝菌纲、优杆菌科、放线菌门、双歧杆菌目、厚壁菌门,fh组中marinifilaceae科、放线菌目、乙型变形菌纲、伯克氏菌科、蓝细菌、gastranaerophilales目、melainabacteria纲、atopobiaceae科显著富集。这些物种在多组中都存在差异,因此对样本的划分产生了显著性差异影响。相比于anova检验方法,lefse的检验方法可以检测到更多的差异性物种,尤其是除属水平之外的差异性显著的物种,可以弥补anova检验方法的空缺。
70.lefse判别分析结果表明,高脂饮食会导致大鼠肠道内某些细菌的显著富集,这些菌属可能与高脂血症的发生呈正相关。经pm治疗后的大鼠肠道菌群在各分类水平上显著富
集的菌属均发生了改变,这可能是其产生治疗作用的关键。
71.综上,本技术采用高通量测序平台对大鼠粪便微生物的16s rrna进行高通量测序,并从微生物的丰富度、多样性、菌群结构和组成变化以及菌群的差异性等方面来考察各组大鼠肠道菌群的变化情况。对粪便样品的样本信息进行统计分析,共得到优化序列数1,337,123条,通过稀释曲线和rank_abundanc曲线分析得知此次各样本的测序深度合理。alpha多样性分析发现,高脂饮食可能会导致肠道菌群物种丰度和多样性减少,而血脂康和不同剂量的pm均可以增加物种丰度和多样性,且pm的效果更优于阳性药。对物种组成变化的分析发现,门水平上,高脂饮食会导致firmicutes/bacteroidetes的比值升高,pm能有效降低这一趋势;属水平上,各分组优势菌属存在差异,fl组和xzk组群落组成最相似;out水平上,经pm治疗后能提高肠道菌群的out组成与正常样本的相似度。β多样性分析发现各组样本组内相似度较高,能与其他组别较好分离;且fh组与nd组在群落构成上最接近。从属水平的物种差异分析来看,高脂饮食导致肠道内微生物群菌属产生较大差异,pm治疗会引起某些菌属丰度的改变,这些变化可能是pm发挥治疗作用的关键。
72.饮食的改变会造成肠道微生物多样性和丰度的改变。长期高脂饮食的大鼠肠道菌群会出现失调现象,其中属水平物种丰度和组成发生了的较大改变,如megamonas属、roseburia属和blautia属丰度的显著增加,高剂量pm可显著上升普雷沃氏菌属的丰度,同时下降muribaculaceae属。最新的研究表明,普雷沃氏菌属/拟杆菌属的比值可以预测饮食干预24周后的体重及脂肪减少,普雷沃氏菌属/拟杆菌属的比值越高,超重受试者摄入高纤维饮食后的体重降低越多。此外,普雷沃氏菌属主要参与碳水化合物和单糖的代谢,能够产生短链脂肪酸,有益于肠道健康。
73.综上,人参/红曲霉发酵产物能有效改善肠道菌群的结构,可能通过特定增加或降低某一菌属水平来实现其功效。使有益菌属增加,有害菌种减少。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。