1.本发明属于人工智能和蛋白质组学领域,具体涉及一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法。
背景技术:
2.胃癌的高发病率和高死亡率反映其诊疗手段不足。目前,曲妥珠单抗仍然是唯一被批准作为her2阳性进展期胃癌患者的一线治疗靶向药,但在全球范围内,胃癌的her2阳性率仅在10.4%到20.2%之间。针对her2阴性的进展期胃癌患者,化疗仍旧是主要的治疗手段。但化疗的总体获益程度是有限的,有的患者能受益于化疗从而获得较好的预后,但有的患者甚至会因化疗的损害而导致更差的预后。显然,并非所有胃癌患者都适合接受化疗,因此区分化疗获益人群对her2阴性胃癌患者的治疗显得尤为重要。
3.为了更好的预测患者预后和指导个体治疗方案,一直以来胃癌分型都是一个热点探索方法。目前基于病理形态学的lauren分型虽然对胃癌治疗起着一定的指导作用,但不同的lauren分型对不同的化疗药物疗效程度差异较大,同种药物对不同的分型疗效亦有差异。随着高通量组学技术的发展,胃癌分型研究的焦点从病理分型转到分子分型。如癌症基因组联盟(tcga)和亚洲癌症研究小组(acrg)基于胃癌的基因组、转录组数据分别定义了四种分子亚型,发现它们具有各自的独特分子特征。如tcga定义的四个亚型分别表现为染色体不稳定性(cin)、微卫星不稳定性(msi)、基因组稳定性(gs)以及epstein
–
barr病毒(ebv)阳性。
4.但是,基因与生命活动的最终执行者—蛋白质之间仍然存在着较大的差异,难以直接反映生命变化。基因水平更多的是反映疾病发生的概率(即可能性),而蛋白质作为基因的效应分子,直接反映了个体的表型,其状态的改变直接地反映了疾病的发生及发展过程。
技术实现要素:
5.为解决以上现有技术存在的问题,本发明提出了一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法,该方法包括:获取待识别的蛋白表达谱,对蛋白表达谱进行预处理;将预处理后的蛋白表达谱输入到训练好的分类器中,得到待识别蛋白表达谱的分类标签;根据分类标签对蛋白表达谱进行分类,完成胃癌蛋白质组学分型框架识别;
6.对分类器进行训练的过程包括:
7.s1:获取患者的蛋白表达谱数据集,并对数据集中的蛋白表达谱进行预处理;
8.s2:采用自动编码器对预处理后的蛋白表达谱进行特征提取,得到亚型标签;
9.s3:采用一致性聚类算法对自动编码器提取的特征进行一致性聚类,得到亚型标签;将所有的亚型标签划分为训练集和测试集,其中训练集用于对分类器进行训练,测试集用于对训练的分类器进行测试;
10.s4:采用单因素cox风险比例模型对训练集中的蛋白进行选择,并对选择后的蛋白进行标准化;
11.s5:将标准化后的蛋白输入到分类器中进行训练,得到训练好的分类器。
12.优选的,对蛋白表达谱进行预处理包括:在蛋白表达谱数据集中筛选出高置信蛋白;剔除高置信蛋白中的高丰度蛋白和冗余蛋白;对经过删除高丰度蛋白和冗余蛋白后的高置信蛋白进行基于综合的定量数据标准化,并去除鉴定频次低于样本总数10%的蛋白,得到标准蛋白表达谱。
13.优选的,采用自动编码器对标准蛋白表达谱进行瓶颈层特征提取的过程包括:将预处理后的蛋白表达谱数据输入自动编码器进行非线性转换,并计算输入数据和重构数据的损失值,当损失值越小时,瓶颈层节点对原始数据的解释性越强,输出此时的瓶颈层节点;采用自动编码器对蛋白表达谱数据进行非线性转换的过程包括:将标准化后的蛋白表达谱输入到自动编码器中,在第一个隐藏层经过relu激活函数转换,得到隐藏节点;将隐藏节点输入到瓶颈层中进行转换,得到转换后的瓶颈节点;将瓶颈节点输入到解码器的第一个隐藏层,并通过relu激活函数转换,得到第一个解码器隐藏层的隐藏节点,将此隐藏节点输入到重构层中,并经过sigmiod函数进行转换,得到自动编码器重构的数据。
14.进一步的,自动编码器的目标函数为:
[0015][0016]
其中,mse表示目标函数,xi表示当前样本的输入数据,xi`表示自动编码器的输出值,n表示样本总数。
[0017]
优选的,采用一致性聚类算法对自动编码器提取出来的特征进行一致性聚类的过程包括:一致性聚类算法为基于欧几里得的k-means聚类算法;设置最大聚类数、重复抽样的比例、抽样次数以及迭代次数;根据设置的重复抽样的比例对样本进行抽样聚类,每次迭代记录样本间的聚类距离;当迭代完成后计算样本间的聚类距离的平均值,将该平均值作为最终的距离矩阵;根据距离矩阵计算样本间的相似性,根据样本相似性计算结果得到聚类结果;聚类结果中每个簇表示亚型标签。
[0018]
优选的,分类器采用随机森林分类器。
[0019]
优选的,采用单因素cox风险比例模型对训练集中的蛋白进行选择的过程包括:将训练集中的蛋白输入到单因素cox风险比例模型中进行风险评估,输出每个蛋白对预后影响的显著性统计值以及风险值;设置阈值,将每个蛋白对预后影响的显著性统计值与设置的阈值进行大小比较,当小于设置的阈值时,则保留该蛋白,否则在训练集中去除该蛋白;单因素cox风险比例模型对蛋白数据进行处理的公式为:
[0020]
h(t)=h0(t)*exp(b1x1 b2x2
…
bnxn)
[0021]
其中,t表示时间,h(t)表示该时间点的死亡风险,x表示因素,exp(bi)表示该因素的hr。
[0022]
为实现上述目的,本发明还提供一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现任一上述基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法。
[0023]
为实现上述目的,本发明还提供一种基于深度学习特征提取的胃癌蛋白质组学分
型框架识别装置,包括处理器和存储器;所述存储器用于存储计算机程序;所述处理器与所述存储器相连,用于执行所述存储器存储的计算机程序,以使所述一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别装置执行任一上述基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法。
[0024]
本发明的有益效果:
[0025]
1)使用深度学习的自动编码器对来自多中心的二期、三期胃癌患者特征提取进行一致性聚类后获得了具有显著生存差异的分子亚型,且亚型表现为预后好、化疗获益和预后差、化疗无效;2)使用随机森林分类器预测出来的患者同样表现为预后好、化疗获益,预后差、化疗无效。
附图说明
[0026]
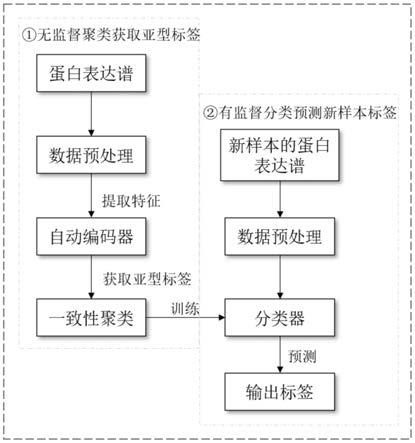
图1为本发明的基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法的流程图;
[0027]
图2为本发明的自动编码器结构图;
[0028]
图3为本发明的通过一致性聚类获取的亚型关联临床信息的k-m曲线示意图;
[0029]
图4为本发明的通过一致性聚类获取的亚型关联化疗状态的k-m曲线示意图;
[0030]
图5为本发明的分类器预测新样本所属亚型生存差异示意图;
[0031]
图6为本发明的利用分类器预测的新样本所属亚型关联化疗状态的k-m曲线。
具体实施方式
[0032]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0033]
本发明主要结合深度学习的自动编码器进行特征提取(降维)用于tumor-node-metastasis分期(tnm stage)为二期、三期的胃癌患者蛋白质组学分子分型。利用自动编码器提取的特征对胃癌样本进行一致性聚类,以期能得到具有生存差异的亚型。除此之外,利用自动编码器提取的特征对胃癌样本进行分型能将化疗获益或者化疗无效甚至化疗有害的人群区分出来。随后,本发明使用基于亚型间独特的特征(如差异蛋白)构建分类器,利用该分类器能为未知所属亚型的样本进行预测,获得样本属于预后好、建议化疗或者预后差、不建议化疗的输出标签,根据预测结果,可以对新样本的治疗进行指导。
[0034]
一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法,如图1所示,该方法包括:获取待识别的蛋白表达谱,对蛋白表达谱进行预处理;将预处理后的蛋白表达谱输入到训练好的自动编码器中进行特征提取,基于一致性聚类获取亚型标签;根据分类标签建立分类器对蛋白表达谱进行分类,完成胃癌蛋白质组学分型框架识别。
[0035]
对分类器进行训练的过程包括:
[0036]
s1:获取患者的蛋白表达谱数据集,并对数据集中的蛋白表达谱进行预处理;
[0037]
s2:采用自动编码器对预处理后的蛋白表达谱进行特征提取;
[0038]
s3:采用一致性聚类算法对自动编码器提取的特征进行一致性聚类,得到亚型标
签;将亚型标签划分为训练集和测试集,其中训练集用于对分类器进行训练,测试集用于对训练的分类器进行测试;
[0039]
s4:采用单因素cox风险比例模型对训练集中的蛋白进行选择,并对选择后的蛋白进行标准化;
[0040]
s5:将标准化后的蛋白输入到分类器中进行训练,得到训练好的分类器。
[0041]
对蛋白表达谱进行预处理包括:在蛋白表达谱数据集中筛选出高置信蛋白;剔除高置信蛋白中的高丰度蛋白和冗余蛋白;对经过删除高丰度蛋白和冗余蛋白后的高置信蛋白进行基于综合的定量数据标准化,并去除鉴定频次低于样本总数10%的蛋白,得到标准蛋白表达谱。
[0042]
如图2所示,自动编码器是一种蝴蝶型对称结构的网络,包含编码器(inputlayer到bottleneck layer部分)和解码器(bottleneck layer的下一层到reconstructedlayer部分)两个部分,编码器主要用于学习输入数据的隐含特征(这个隐含特征表现为瓶颈层的节点),解码器主要是用编码器获得的隐含特征重构出输入数据。
[0043]
采用自动编码器对标准蛋白表达谱进行瓶颈层特征提取的过程包括:将预处理后的蛋白表达谱数据输入自动编码器进行非线性转换,并计算输入数据和重构数据的损失值,当损失值越小时,瓶颈层节点对原始数据的解释性越强,输出此时的瓶颈层节点;采用自动编码器对蛋白表达谱数据进行非线性转换的过程包括:将标准化后的蛋白表达谱输入到自动编码器中,在第一个隐藏层经过relu激活函数转换,得到隐藏节点;将隐藏节点输入到瓶颈层中进行转换,得到转换后的瓶颈层节点;将瓶颈层节点输入到解码器的第一个隐藏层,并通过relu激活函数转换,得到解码器第一个隐藏层的隐藏节点,将此隐藏节点输入到重构层中,并经过sigmiod函数进行转换,得到自动编码器重构的数据。
[0044]
一种采用自动编码器对标准蛋白表达谱进行瓶颈层特征提取的具体实施方式,包括将预处理后的蛋白表达谱数据输入自动编码器进行非线性转换,计算输入数据和重构数据的损失值,当损失值越小时,瓶颈层节点对原始数据的解释性越强,输出此时的瓶颈层节点。首先,将标准化后的蛋白表达谱输入自动编码器,在第一个隐藏层经过relu激活函数转换(在这里,小于0的值全部转换为0,大于0的值保持其值不变),最后在第一个隐藏层输出500个隐藏节点进入瓶颈层,瓶颈层将这500个节点转换为100个节点。接下来,将经过瓶颈层转换后的100个节点输入到解码器的第一个隐藏层,同样结果经过relu激活函数转换,输出500个节点到重构层。最后,重构层输出与输入表达谱相同维度并经过sigmiod转换的节点。
[0045]
将预处理后的胃癌蛋白表达谱进行标准化,作为自动编码器的输入,取自动编码器瓶颈层节点作为新特征。自动编码器隐藏层数为3层,节点数分别为500,100,500。隐藏层选用relu作为其激活函数,但瓶颈层不设置任何激活函数。relu激活函数计算公式如下:
[0046]
y=f(x)=max(0,x)
[0047]
其中,x表示当前样本的输入数据。
[0048]
对于重构层,选用sigmoid作为其激活函数,激活函数的表达式为:
[0049][0050]
对于自动编码器网络,选用均方误差(mse)作为其目标函数(损失函数),用以评估
输入x和输出x’之间的误差。mse计算公式如下:
[0051][0052]
其中,mse表示目标函数,xi表示当前样本的输入数据,x`i表示自动编码器的输出值,n表示样本总数。
[0053]
将自动编码器瓶颈层的节点提取出来之后,使用r包——consensusclusterplus执行一致性聚类。聚类算法采用基于欧几里德距离的k-means。每次重采样选取的样本比例为80%。具体的,一致性聚类算法为基于欧几里得的k-means聚类算法;设置最大聚类数5、重复抽样的比例80%以及抽样次数200次;根据设置的重复抽样的比例对样本进行抽样聚类,每次迭代记录样本间的聚类距离;当迭代完成后计算样本间的聚类距离的平均值,将该平均值作为最终的距离矩阵;根据距离矩阵计算样本间的相似性,根据样本相似性计算结果得到聚类结果;聚类结果中每个簇表示亚型标签。
[0054]
采用kaplan-meier方法对一致性聚类获得的亚型与患者对应的临床信息进行关联分析,说明亚型间的预后差异以及患者对化疗药物的响应情况(化疗获益、化疗无效、化疗有害)。
[0055]
采用单因素cox风险比例模型对训练集中的蛋白进行选择的过程包括:将训练集中的蛋白输入到单因素cox风险比例模型中进行风险评估,输出每个蛋白对预后影响的显著性统计值以及风险值;设置阈值,将每个蛋白对预后影响的显著性统计值与设置的阈值进行大小比较,当小于设置的阈值时,则保留该蛋白,否则在训练集中去除该蛋白;单因素cox风险比例模型对蛋白数据进行处理的公式为:
[0056]
h(t)=h0(t)*exp(b1x1 b2x2
…
bnxn)
[0057]
其中,t表示时间,h(t)表示该时间点的死亡风险,x表示因素,exp(bi)表示该因素的hr。(hr=1:无影响,hr<1:降低风险,hr>1,增加风险)利用python的coxphfitter函数计算每个蛋白的风险值及其p值,最后输出格式如下的统计矩阵(部分统计结果):
[0058] coefexp(coef)p-log2(p)lower0.95upper 0.95protein0.0001041.0001040.9774970.032835-0.0071230.007331
[0059]
其中,exp(coef)即为该蛋白的hr,p即为该蛋白对预后的显著性统计值,lower和upper分别为hr的置信区间。
[0060]
本发明的一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法中的数据来自多中心的833例二期、三期胃癌福尔马林固定、石蜡包埋(ffpe)的手术切除样本。首先将这833例样本划分为发现集和独立验证集,再按7:3的比例将发现集随机划分为训练集和测试集。利用该框架发现在发现集中约占43%的患者受益于辅助化疗,这组患者的预后显著优于那些未受益于化疗的患者。即预后较好的亚型接受化疗的患者五年生存率比不接受化疗的患者五年生存率提高了12个百分点,预后较差的亚型接受化疗的患者和不接受化疗的患者五年生存率没有显著改善。
[0061]
随后,利用两个亚型的分子特征构建分类器,在训练集上结合10折交叉验证训练分类器,并在测试集测试分类器性能,最后在独立验证集验证通过自动编码器提取特征进行分子分型预测胃癌患者预后及化疗获益人群的性能稳定性。最后,通过构建分类器对独立验证集的样本进行亚型预测,输出独立验证集样本对应的亚型标签,关联临床信息。我们
发现,独立验证集样本预测的所属亚型标签同样具有生存差异,且预后好的亚型接受化疗的患者五年生存率比不接受化疗的患者提高了25个百分点,同样预后较差的亚型接受化疗的患者同未接受化疗的患者相比没有显著差异。
[0062]
如图3所示,本发明通过自动编码器提取特征进行分型能够将胃癌患者预后不同的人群区分开。预后主要关心病人存活状态以及存活时间,通过绘制k-m曲线能比较直观的展示亚型的一个预后情况。由图可以获得的信息是亚型一病人较亚型二预后良好,术后存活时间较长。
[0063]
由于本发明除了需要将不同预后的病人区分开外,还希望能将化疗获益的人群也区分出来,而根据临床表现期望的应该是病人因受到化疗疗效的作用产生较好的预后,或者说病人因受到化疗毒性的副作用而产生更差的预后,因此,在这还将亚型单独关联化疗状态分析,同样通过k-m曲线直观的展示病人化疗后的预后情况。图4左为亚型一病人关联化疗状态的生存曲线,由图可以看出亚型一中接受化疗的患者较未接受化疗的患者生存率具有显著的提高,接受化疗的患者五年生存率为65.3%,未接受化疗的患者为52.6%,整体提高12个百分点;图4右为亚型二病人关联化疗状态的生存曲线,其中接受化疗的患者五年生存率为54.2%,未接受化疗患者为51.1%,没有显著提高。
[0064]
发现集上通过一致性聚类获取的亚型最终需要建立分类器进行预测,只有分类器预测样本同样表现为亚型一预后良好、化疗获益,亚型二预后不良、化疗无效才能说明结果前述获取的亚型可靠,该方法整体可靠。因此图5图6基本同图4图5,只是图5-6是预测集上的结果。
[0065]
于本发明一实施例中,本发明还包括一种计算机可读存储介质,其上存储有计算机程序,该程序被处理器执行时实现上述任一所述基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法。
[0066]
本领域普通技术人员可以理解:实现上述各方法实施例的全部或部分步骤可以通过计算机程序相关的硬件来完成。前述的计算机程序可以存储于一计算机可读存储介质中。该程序在执行时,执行包括上述各方法实施例的步骤;而前述的存储介质包括:rom、ram、磁碟或者光盘等各种可以存储程序代码的介质。
[0067]
一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别装置,包括处理器和存储器;所述存储器用于存储计算机程序;所述处理器与所述存储器相连,用于执行所述存储器存储的计算机程序,以使所述一种基于深度学习特征提取的胃癌蛋白质组学分型框架识别装置执行任一上述基于深度学习特征提取的胃癌蛋白质组学分型框架识别方法。
[0068]
具体地,所述存储器包括:rom、ram、磁碟、u盘、存储卡或者光盘等各种可以存储程序代码的介质。
[0069]
优选地,所述处理器可以是通用处理器,包括中央处理器(central processing unit,简称cpu)、网络处理器(network processor,简称np)等;还可以是数字信号处理器(digital signal processor,简称dsp)、专用集成电路(application specific integrated circuit,简称asic)、现场可编程门阵列(field programmable gate array,简称fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件。
[0070]
以上所举实施例,对本发明的目的、技术方案和优点进行了进一步的详细说明,所应理解的是,以上所举实施例仅为本发明的优选实施方式而已,并不用以限制本发明,凡在
本发明的精神和原则之内对本发明所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
