1.本发明涉及化学药物中间体技术领域,具体涉及一种帕西洛韦(paxlovid)中间体的合成方法。
背景技术:
2.化合物(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐用式(a)表示,是合成新冠抗病毒口服药帕西洛韦(paxlovid)和丙型肝炎蛋白酶抑制剂波普瑞韦重要的中间体,由(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐合成帕西洛韦,具有操作简单、利于工业化生产等特点。该中间体结构式如下:
[0003][0004]
pct国际专利申请wo2004/113295公开了一种(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐的合成方法,其具体合成路线如下:
[0005][0006]
其中,r2为氢或乙烷。虽然该方法合成了帕西洛韦中间体(s)-3-氨基-n-环丙基-2-羟基己酰胺盐酸盐,但是该合成过程中使用昂贵的钯碳催化剂和比较危险的氢化铝锂还原剂,在工业化生产中安全性是很大的挑战,而且反应需要使用特殊设备,导致生产成本较高,能耗和设备的占用率高。此外,该合成方法的步骤极其繁琐,反应时间较长,不仅增大了生产成本和后处理成本,还严重影响最终产物的收率和纯度。
[0007]
pct国际专利申请wo2007/075790公开了一种(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐的合成方法,其具体合成路线如下:
[0008]
虽然该方法合成过程中同样使用了昂贵的催化剂钯碳、箔碳和agno3,和采用了强氧化剂过硫酸钾和危险的氢化铝锂还原剂。反应时对设备需要有特殊要求,此外该合成方法的步骤繁琐,反应时间较长,如最后一步就需要在烘干箱中处理3天左右,该方法对化合物的手性无法控制,需要手性拆分,增加操作步骤,还严重影响产品的收率和生产成本。
[0009]
中国专利公开号cn103435532b公开了一种(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐的合成方法,其具体合成路线如下:
[0010][0011]
该方法采用6,6-二甲基-3-氮杂双环[3.1.0]己烷盐酸盐为原料,难以获得,价格高。反应中使用了极易燃的仲丁基锂,危险性较高。最后一步使用了腐蚀性较大的二氯亚砜,对设备有较高的防腐要求。该路线并无手性控制的策略,通过手性拆分,增加操作步骤,还严重影响产品的收率和生产成本。
[0012]
因此,针对现有技术中存在的上述问题,本发明提供一种原料低价易得、能耗低、制备简单、安全性高且降低因手性拆分导致的损失的制备方法。
技术实现要素:
[0013]
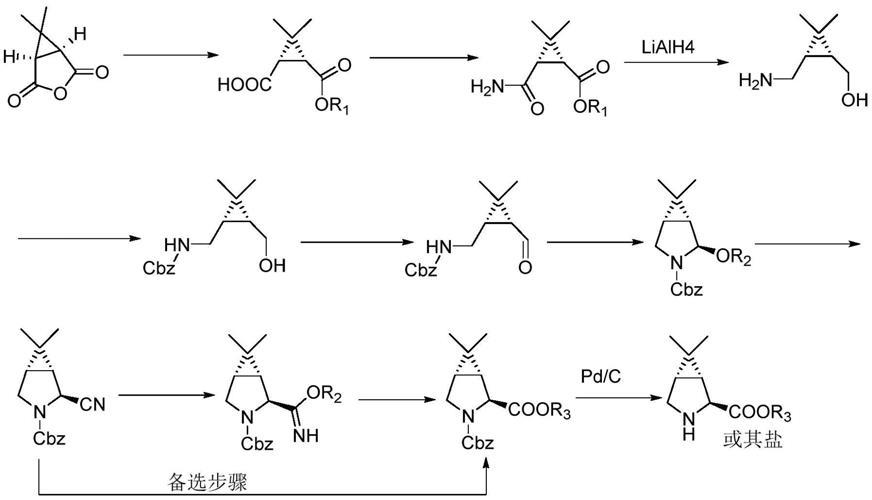
本发明的目的是提供(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯的制备方法,以3,3-二甲基-4-氧代丁酸乙酯为起始原料,引入s-叔丁基亚磺酰胺基团,
在合成桥环结构的过程中,s-叔丁基亚磺酰胺基团能够诱导作用,选择性合成所需的对映异构体,避免了现有合成方法中因手性拆分导致的损失。
[0014]
为实现上述发明目的,本发明的技术方案如下:
[0015]
一种(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯的制备方法,包括如下步骤:
[0016]
(1)3,3-二甲基-4-氧代丁酸酯与s-取代亚磺酰胺反应生成化合物c-1;
[0017]
(2)化合物c-1和单卤素取代乙酸酯在强碱作用下反应,利用s-取代亚磺酰胺手性诱导,得到化合物c-2;
[0018]
(3)化合物c-2在强碱作用下形成过渡态中间体化合物c-3,进一步在强碱作用下和s-取代亚磺酰手性诱导下,生成化合物c-4;
[0019]
(4)化合物c-4脱保护基得化合物c-5;
[0020]
(5)化合物c-5经还原反应得(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯,
[0021][0022]
优选地,r1、r3各自独立的选自脂肪烃或芳香烃;进一步优选为烷基,最优选为乙基。
[0023]
优选地,r2选自烷基、芳基、取代芳基中的任一种;进一步优选为c2-6烷基、苯基、取代苯基中的任一种;更进一步优选为丁基,最优选为叔丁基。
[0024]
优选地,步骤(1)中,需要加入催化剂,所述催化剂选自硫酸铜,四氯化钛,钛酸四甲酯,钛酸四乙酯,钛酸四异丙酯,钛酸四丁酯,氯化锌,氯化铝中的一种或几种;最优选为钛酸四乙酯。
[0025]
优选地,步骤(2)中,所述单卤素取代乙酸酯中的卤素为氯或溴,进一步优选为氯。
[0026]
优选地,步骤(2)中,所述强碱选自叔丁醇钠,叔丁醇钾,乙醇钠,乙醇钾,甲醇钠,甲醇钾,lda,正丁基锂,甲醇锂,叔丁醇锂,lihmds中的一种;最优选为lihmds。
[0027]
在步骤(2)中,利用强碱拔去单卤素取代的乙酸酯上的氢,加成到中间体c-1的碳氮双键上,形成氮杂三元环,并利用s-取代亚磺酰胺手性诱导三元环上两个叔碳的手性。
[0028]
优选地,步骤(3)中,所述强碱选自叔丁醇钠,叔丁醇钾,乙醇钠,乙醇钾,甲醇钠,甲醇钾,lda,正丁基锂,甲醇锂,叔丁醇锂,lihmds中的一种;最优选为叔丁醇钠。
[0029]
在步骤(3)中,中间体c-2在强碱拔氢作用下,氮杂三元环打开,环丙烷化形成过渡
态中间体c-3,进一步在强碱作用下关上五元环,并利用s-取代亚磺酰手性诱导作用,控制五元环上的立体构型。
[0030]
优选地,步骤(4)中,所述脱保护基通过加酸实现,进一步优选的,所述酸选自硫酸,盐酸,磷酸,三氟乙酸,甲磺酸,对甲苯磺酸中的一种或几种;最优选为盐酸。
[0031]
优选地,步骤(5)中,所述还原反应需要加入还原剂,所述还原剂选自氢化锂铝,硼烷,硼氢化钠中的一种;最优选为硼氢化钠。
[0032]
优选地,步骤(5)中,(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯再经盐酸成盐步骤得到(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯盐酸盐。
[0033]
本发明的有益效果为:
[0034]
(1)所用原料为3,3-二甲基-4-氧代丁酸酯,原料廉价,合成简单易操作;所用的试剂均为常规试剂,并未使用昂贵的以及需要特殊设备的试剂,能耗低,生产成本低;
[0035]
(2)利用s-取代亚磺酰手性诱导作用,实现手性控制,在合成桥环结构的过程中,s-叔丁基亚磺酰胺基团能够诱导作用,选择性合成所需的对映异构体。避免了现有合成方法中因手性拆分导致的损失;
[0036]
(3)该方法制备的(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸酯立体选择性高、收率好。
具体实施方式
[0037]
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例,进一步阐明本发明,但下述实施例仅为本发明的优选实施例,并非全部。基于实施方式中的实施例,本领域技术人员在没有做出创造性劳动的前提下所获得其它实施例,都属于本发明的保护范围。下述实施例中,若无特殊说明,所用的操作方法均为常规操作方法,所用设备均为常规设备,各个实施例所用设备材料均相同。
[0038]
下述实施例中,所用原料3,3-二甲基-4-氧代丁酸乙酯由文献(reissig h u,reichelti,kunz t.methoxycarbonylmethylation of aldehydes via siloxycyclopropanes:methyl 3,3-dimethyl-4-oxobutanoate[m].john wiley&sons,inc.2003.)所述方法合成。
[0039]
本发明制备路径如下:
[0040][0041]
实施例1中间体c-1的制备
[0042]
将3,3-二甲基-4-氧代丁酸乙酯(10.3g,65mmol)溶于100ml二氯甲烷中,加入叔丁基亚磺酰胺(15.6g,129mmol),滴加钛酸四乙酯(17.6g,77mmol),25℃搅拌反应,tlc监测反应进程。反应结束后,向反应瓶内加入100ml水淬灭反应。过滤,滤液分层,弃水相。有机相浓缩,硅胶柱层析纯化得15.47g中间体c-1,纯度95.3%,收率91.1%。
[0043]1h-nmr(cdcl3,400mhz):δ4.11(q,j=7.2hz,2h),2.62-2.50(m,2h),1.34(s,3h),1.32(s,3h),1.29(t,j=7.2hz,3h),1.25(s,9h)ppm.
[0044]
实施例2中间体c-2的制备
[0045]
向100ml三口瓶中加入氯乙酸乙酯(1.5g,12.5mmol),氮气置换三次,加入干燥的四氢呋喃40ml,冷却至-78℃。缓慢滴加1m双三甲基硅基胺基锂四氢呋喃溶液(12.5ml,12.5mmol),-78℃搅拌十分钟。中间体c-1(1.3g,5mmol)溶解于3ml四氢呋喃中,缓慢注射至反应液中。加料完毕后-78℃搅拌2小时。向反应液加水淬灭,乙酸乙酯萃取,干燥、浓缩有机相,硅胶柱层析纯化得到1.55g浅黄色油状物c-2,纯度91.2%,收率89.3%。
[0046]1h-nmr(cdcl3,400mhz):δ4.30-4.11(m,4h),2.90-2.82(m,2h),2.37-2.23(m,2h),1.40(s,9h),1.36(s,3h),1.32(s,3h),1.31-1.22(m,6h)ppm.
[0047]
实施例3中间体c-4的制备
[0048]
向100ml单口瓶中加入30ml四氢呋喃,中间体c-2(1.7g,5mmol)和叔丁醇钠(960mg,10mmol)室温搅拌3小时。向反应液加水淬灭,乙酸乙酯萃取,干燥、浓缩有机相,硅胶柱层析纯化得到1.1g浅黄色油状物c-4,纯度92.4%,收率73.3%。
[0049]1h-nmr(cdcl3,400mhz):δ4.16(q,j=7.2hz,2h),3.13-3.06(m,1h),2.21-2.12(m,1h),1.62-1.54(m,1h),1.36(s,9h),1.32(t,j=7.2hz,3h),1.25(s,3h),1.23(s,3h)ppm.
[0050]
实施例4中间体c-5的制备
[0051]
将中间体c-4(0.3g,1mmol)溶解于3ml 1,4-二氧六环中,加入1m氯化氢的1,4-二氧六环溶液(2ml,2mmol),室温搅拌3小时。加入碳酸氢钠溶液中和,乙酸乙酯萃取。干燥、浓缩有机相,硅胶柱层析纯化得到177mg中间体c-5,纯度94.9%,收率89.8%。
[0052]1h-nmr(cdcl3,400mhz):δ4.11(q,j=7.2hz,2h),3.22-3.11(m,1h),2.23-2.15(m,
1h),1.66-1.59(m,1h),1.34(t,j=7.2hz,3h),1.26(s,3h),1.23(s,3h)ppm.
[0053]
实施例5中间体c-6的制备
[0054]
将中间体c-5(2g,10mmol)溶解于30ml乙醇中,加入硼氢化钠(450mg,12mmol),加热回流搅拌7小时。稀盐酸调节至中性,乙酸乙酯萃取,有机相浓缩干。浓缩物溶解于1ml 30%氯化氢乙醇溶液,加入10mlmtbe,室温搅拌2小时,析出大量固体。过滤、干燥得到1.9g白色固体,收率88.3%,纯度99.1%,ee值大于97%。
[0055]1h-nmr(cd3od,400mhz):δ4.31(d,j=1.6hz,1h),4.20(t,j=7.2hz,2h),3.77-3.74(m,1h),3.31-3.28(m,1h),2.00-1.94(m,1h),1.84-1.79(m,1h),1.33(t,j=7.2hz,3h),1.21(s,3h),1.18(s,3h)ppm.
[0056]
实施例6
[0057]
与实施例2不同的是,将实施例2中的双三甲基硅基胺基锂替换为相同用量的叔丁醇钠,其余皆相同,纯度88.4%,收率42.3%。
[0058]
实施例7
[0059]
与实施例3不同的是,将实施例3中的叔丁醇钠替换为相同用量的双三甲基硅基胺基锂,其余皆相同,纯度90.2%,收率22.5%。
[0060]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。