1.本发明属于医学材料技术领域,具体涉及一种低肾毒性蛋白-氧化铁复合纳米磁共振造影剂及其制备方法和应用。
背景技术:
2.磁共振成像(mri)技术由于无创、无辐射成为当前医学诊断的有力工具,为了达到更好的成像效果,目前约有40-50%造影剂用于mri检测。磁共振造影剂通常根据对纵向或横向弛豫产生的影响分为纵向(t1)造影剂和横向(t2)造影剂两类。
3.具有正增强效果的t1造影剂更适合于高分辨率成像,目前临床上广泛使用的t1造影剂是钆基造影剂(gbca)。然而,gbca在临床应用中有几个缺点:gbca复合物通常通过尿液快速排出,这妨碍了它们用于需要较长扫描时间的高分辨率成像。此外,已知它们具有毒性副作用,由钆络合物中游离的gd
3
离子引起,包括肾源性系统纤维化(nsf)并伴有肾功能损害。另外,钆可在患者反复接触gbca后积聚在骨骼甚至脑组织中,并在这些器官中保留多年。gbca造影剂的这些缺点极大限制了此类造影剂在临床中的应用。近年来,已经对gbca标准的临床应用替代品进行了深入的研究,逐渐重视基于纳米氧化铁的mri造影剂的开发。
4.由于铁的普遍分布,氧化铁比钆基材料更具生物相容性。近年来,超顺磁性氧化铁纳米颗粒(spion)在生物医学应用中引起了极大的兴趣。然而,目前通常通过热分解和水热法合成小尺寸的spion,这些方法通常比较复杂,对环境有害且耗时长。另外,生产spion的过程通常涉及使用有机溶剂,导致spion具有疏水区域,可能造成严重的非特异性效应。虽然在spion上更换配体可以赋予其亲水性和生物相容性,但无疑增加了亲水配体置换的复杂性,降低了合成效率。此外,现有spion的常用亲水配体多数为高分子量peg(多聚乙二醇)或者多聚糖(例如fad通过的ferumoxytol),在体内难以降解,导致其材料无法被肾脏快速清除,潜在肾脏毒性较大,限制了其临床应用。
5.将天然蛋白质引入超小氧化铁纳米颗粒的合成过程,无疑是一个更好的解决办法。铁蛋白ferritin是一种普遍存在的储铁蛋白,它可以组装成一个纳米笼状结构,外径12nm,内腔8nm,目前被大量研究用于超小尺寸氧化铁纳米颗粒的合成。然而,ferritin蛋白产量有限、成本较高,因而无法用于氧化铁纳米颗粒的大量制备,并且目前报道的ferritin介导合成的氧化铁纳米颗粒多为t2造影剂,t1造影效果不佳。相比之下,牛血清白蛋白(bsa)具有更大的优势,其价格低廉,并且易于进行功能化修饰,已被大量用于金属氧化物纳米颗粒的合成。然而,以bsa模板合成用于磁共振血管造影(mra)的氧化铁纳米颗粒仍是一个很大的挑战。
6.cn110787307a公开一种磁共振成像纳米造影剂及其制备方法和应用,该文献方法首先在回流的条件下制得fe2o3超级纳米粒子(fe2o
3 sps),然后将牛血清白蛋白bsa吸附至fe2o
3 sps的表面以得到bsa修饰的fe2o
3 sps,该bsa修饰的fe2o
3 sps具有优异的磁共振成像效果。然而该文献并没有验证其制备的磁共振成像纳米造影剂的造影效果,且制备方法
分为先制备fe2o
3 sps,随后再以bsa包裹fe2o
3 sps,步骤繁琐,得到的磁共振成像纳米造影剂的无机核心粒径较大,不易被吸收,通过肾脏较难清除,限制了其血管造影效果和生物安全性。
技术实现要素:
7.针对现有技术中存在的问题的一个或多个,本发明提供一种低肾毒性蛋白-氧化铁复合纳米磁共振造影剂及其制备方法和应用。
8.本文中术语“功能化蛋白”是指富含羧基、巯基氨基酸等酸性氨基酸的蛋白质,可以与二价三价铁离子发生螯合作用,并且在铁离子共沉淀反应中,可以形成多聚体状态的蛋白纳米笼子,包覆在无机纳米材料(例如fe2o3、fe3o4等)的表面使其具有很好的亲水性与单分散性。同时这类蛋白可以使纳米材料具有更好的生物兼容性与更长血液循环周期。这类功能化蛋白的实例包括bsa(牛血清白蛋白,其具有延长药物半衰期,并可提高药物的生物兼容性)、hsa(人血清白蛋白,其具有延长药物半衰期,并提高药物的人体生物兼容性及安全性)和h-ferritin(人铁蛋白,其具有延长药物半衰期,并提高药物的人体生物兼容性及安全性,同时具有靶向到肿瘤组织的作用),还包括dna-binding proteins(dna结合蛋白,其具有延长药物半衰期,并可提高药物的生物兼容性)、heart-shock protein(热休克蛋白,其具有延长药物半衰期,并可提高药物的生物兼容性,同时具有靶向到肿瘤组织的作用)。
9.在本发明的一个方面,提供一种低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的制备方法,其包括以下步骤:
10.1)将功能化蛋白溶液与铁盐溶液中混合,得到第一混合溶液;
11.2)向第一混合溶液中加入naoh溶液混合反应,获得第二混合溶液;
12.3)过滤第二混合溶液后,经分离纯化获得低肾毒性蛋白-氧化铁复合纳米磁共振造影剂。
13.上述方法中,步骤1)和步骤2)均在惰性气体保护下进行。
14.上述方法在步骤2)之后还包括步骤2a):向所述第二混合溶液中加入过量的过氧化氢溶液混合反应;随后执行步骤3)获得低肾毒性蛋白-氧化铁复合纳米磁共振造影剂。
15.上述方法中,步骤1)、步骤2)和步骤2a)均在水浴和搅拌条件下进行,其中水浴温度为25~39℃,搅拌的转速为200~600转/min。
16.上述方法中,步骤1)中所述功能化蛋白选自以下中的任一种或其混合:牛血清白蛋白、人血清白蛋白、人铁蛋白、dna结合蛋白和热休克蛋白。
17.上述方法中,步骤1)中所述铁盐溶液为fe
2
与fe
3
的混合溶液,其中fe
2
与fe
3
的摩尔比为1:4~4:1。
18.上述方法中,步骤1)中所述第一混合溶液中fe与功能化蛋白的摩尔比为20:1~660:1,优选为20:1~200:1,进一步优选为20:1~165:1。
19.上述方法中,步骤1)中所述第一混合溶液中还含有保护试剂,所述保护试剂选自以下中的任一种或其混合:peg及其衍生物、尿素、柠檬酸、edta、氨基酸;优选所述保护试剂在第一混合溶液中的浓度不超过50mg/ml。
20.上述方法中,步骤2)中所述naoh溶液的加入量为使得溶液中的所有fe
2
与fe
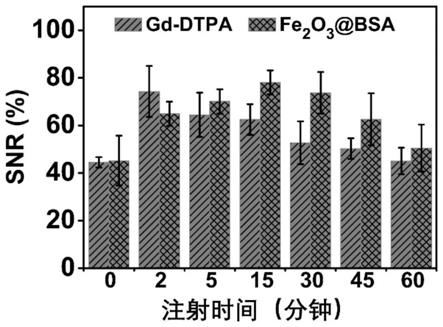
3
均
沉淀的量,例如使得溶液中naoh与fe离子的总量的摩尔比大于2:1,优选大于3:1。
21.上述方法中,步骤2)中所述向第一混合溶液中加入naoh溶液的速率为10ml/h~15ml/h。
22.上述方法中,步骤3)中所述分离纯化的具体操作为:将过滤后的溶液稀释到生理盐水或磷酸缓冲液中,透析后收集透析液;随后浓缩所述透析液,之后用分子筛从浓缩液中分离纯化获得低肾毒性蛋白-氧化铁复合纳米磁共振造影剂。
23.在本发明的另一方面,提供一种低肾毒性蛋白-氧化铁复合纳米磁共振造影剂,其由上述的方法制备。
24.上述低肾毒性蛋白-氧化铁复合纳米磁共振造影剂以氧化铁为核心,功能化蛋白分子包裹于该核心氧化铁上,其中所述核心的粒径为2.94nm~8.04nm,优选为2.94nm~5.50nm。
25.上述氧化铁选自以下中的任一种或其混合:fe2o3、fe3o4;所述功能化蛋白选自以下中的任一种或其混合:牛血清白蛋白、人血清白蛋白、人铁蛋白、dna结合蛋白和热休克蛋白。
26.上述低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的亲水合粒径为15nm~55nm,优选为15nm~40nm,1.5t到3t磁场范围内的r1值为4.5-18mm-1
s-1
,1.5t到3t磁场范围内的r2/r1的比值为5到16.5。
27.上述的低肾毒性蛋白-氧化铁复合纳米磁共振造影剂在非医用磁共振成像中的应用也属于本发明的内容。
28.基于以上技术方案提供的低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的制备方法通过控制反应体系中的功能化蛋白与铁离子的比例关系,并控制反应程序和条件,可以利用功能化蛋白自组装笼状生物模板合成具有高度均一性和单分散性的超小尺寸氧化铁(例如fe2o3、fe3o4或其混合)纳米粒子,进而获得以该超小尺寸氧化铁纳米粒子为无机核心,功能化蛋白分子包裹于其上的蛋白-氧化铁复合纳米磁共振造影剂(即本文所述氧化铁@bsa纳米颗粒),该制备方法简单、原料易得,一锅原位法(一步法)即可获得具有高度均匀性、亲水性和单分散性优势的蛋白-氧化铁复合纳米磁共振造影剂,合成效率高且成本低。并且制备获得的蛋白-氧化铁复合纳米磁共振造影剂的无机核心氧化铁纳米粒子的尺寸较小,达到2.94nm~8.04nm,因此在体内容易被吸收代谢清除,潜在肾脏毒性较小;且在临床强度施加的磁场下显示出较低的r1值和饱和磁化强度,有利于增强作为造影剂(例如t1造影剂)的对比效果。实施例结果表明,本发明提供的氧化铁@bsa纳米颗粒(例如fe2o3@bsa纳米颗粒)作为t1造影剂时具有与市售t1造影剂gd-dtpa相当的t1加权mra效应(即可以提高病灶区域的对比的snr最大效果达到约70%),但相对于目前临床医用的gd-dtpa造影剂具有更亮信号,更长延长时间的血管造影效果,更有利于稳态和高分辨率成像。体外和体内生物稳定性、毒性和肾脏清除率评估结果显示本发明提供的造影剂具有良好的生物相容性和生物安全性,在合适的剂量下,可以很容易地消除肾脏损伤,无明显的副作用,显示出作为一种安全的低肾毒性磁共振造影剂(例如t1造影剂)的巨大潜力和应用价值。
附图说明
29.图1为实施例1中制备低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的装置结构示
意图;
30.图2为实施例1中纳米颗粒fe2o3δbsa(即纳米颗粒fe2o3)的表征图像,其中a幅为tem图像,b幅为尺寸分布柱状图;
31.图3为实施例1中纳米颗粒fe2o3@bsa4(即图中的fe2o3@bsa,以下图中同)的表征图像,其中a幅和b幅为tem图像,c幅为xrd图谱,d幅为fe2o3@bsa4纳米颗粒中无机核心fe2o3的尺寸分布图,e幅为fe2o3@bsa4纳米颗粒的亲水合粒径检测结果,f幅为拉曼光谱图像,g和h幅为经负染的fe2o3@bsa4纳米颗粒的tem图像,i幅为蛋白经分子筛洗脱曲线;
32.图4为实施例1中纳米颗粒fe3o4@bsa的表征图像,其中a幅为tem图像,b幅为纳米颗粒fe3o4@bsa中无机核心fe3o4的尺寸分布;
33.图5为实施例1中纳米颗粒fe2o3@bsa4的ftir光谱;
34.图6为实施例1中纳米颗粒fe2o3@bsa4的xps光谱;
35.图7为标准蛋白的洗脱曲线;
36.图8为经纯化的纳米颗粒fe2o3@bsa4的sds-page凝胶电泳图像;
37.图9为fe2o3@bsa4纳米颗粒与gd-dtpa的磁性能对比结果,其中a幅为在300k时与磁场有关的磁化曲线(m-h)和矫顽力(hc),b幅为在100oe下零场冷却(zfc)和场冷却(fc)后的温度相关磁化曲线(m-t),c幅为t1弛豫率图,d幅为t2弛豫率图,e幅和f幅分别示出了t1加权mr图像和t2加权mr图像;
38.图10为fe3o4@bsa纳米颗粒的磁性能检测结果曲线,其中a幅为在300k时与磁场有关的磁化曲线(m-h),b幅为在100oe下零场冷却(zfc)和场冷却(fc)后的温度相关磁化曲线(m-t);
39.图11为fe3o4@bsa纳米颗粒的t1和t2弛豫率以及t1和t2加权mr图像,其中a幅为t1和t2弛豫率,b幅为t1和t2加权mr图像;
40.图12为fe2o3@bsa4纳米颗粒的体内mr造影结果,其中a幅为在3t场强下注射fe2o3@bsa4纳米颗粒和gd-dtpa后,大鼠的t1加权mr图像的比较,b幅为注射后心脏的信噪比(snr)的对比度增强比,c幅表示在3t下注射fe2o3@bsa4纳米颗粒的大鼠的t1加权mr血管造影(mra)的3d图像;
41.图13为注射前和注射后心脏的信噪比(snr);
42.图14为fe2o3@bsa4纳米颗粒的体外稳定性、安全性、生物相容性和体内分布的时间依赖性评价结果,其中a幅为fe2o3@bsa4纳米颗粒在pbs中储存不同时间的cd光谱,b幅为fe2o3@bsa4纳米颗粒的时间依赖性hd变化曲线及照片,c幅为fe2o3@bsa4纳米颗粒在不同fe浓度下的hd变化曲线及照片,d幅为培养在含有不同fe浓度纳米颗粒培养基中的hepg2和293t细胞的cck-8细胞活力测定结果柱状图,e幅为fe2o3@bsa4纳米颗粒的溶血评价曲线及照片,f幅为尾静脉注射fe2o3@bsa4纳米颗粒后24h和48h,大鼠主要器官中fe元素的时间依赖性生物分布柱状图;
43.图15为fe2o3@bsa4纳米颗粒在不同ph下的hd变化曲线及照片;
44.图16为注射fe2o3@bsa4纳米颗粒7天和14天后收获的大鼠主要器官的h&e组织病理染色图像;
45.图17为fe2o3@bsa4纳米颗粒注射前、注射后2h、4h、6h、12h、24h、48h大鼠尿液中的fe含量变化柱状图;
46.图18中a幅和b幅分别为注射fe2o3@bsa4纳米颗粒后2h和24h大鼠尿液样本的tem图像,c幅为注射fe2o3@bsa4纳米颗粒后大鼠血液生化指标变化柱状图。
具体实施方式
47.以下结合具体实施例,对本发明进一步阐述。应当理解的是,具体实施例仅用于进一步说明本发明,而不是用于限制本发明的内容。
48.下述实施例中所用方法如无特别说明均为常规方法。
49.实施例中描述到的各种生物材料的取得途径仅是提供一种实验获取的途径以达到具体公开的目的,不应成为对本发明生物材料来源的限制。事实上,所用到的生物材料的来源是广泛的,任何不违反法律和道德伦理能够获取的生物材料都可以按照实施例中的提示替换使用。
50.以下实施例中以制备牛血清白蛋白(bsa)包裹fe2o3的低肾毒性蛋白-氧化铁复合纳米磁共振造影剂以及牛血清白蛋白包裹fe3o4的低肾毒性蛋白-氧化铁复合纳米磁共振造影剂为例说明,其中使用的材料及其来源:亚铁和铁盐(feso4·
7h2o和fecl3·
6h2o)、bsa、过氧化氢(h2o2)和氢氧化钠(naoh)均购自sigma-aldrich。活/死细胞染色试剂盒和细胞计数试剂盒-8均购自上海贝斯特比。magnevist(马根维显,gd-dtpa类造影剂)购自中国北京北陆制药有限公司。
51.实施例1:低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的制备及表征
52.1)低肾毒性蛋白-氧化铁复合纳米磁共振造影剂的制备
53.该实施例采用化学共沉淀法制备了多种具有不同fe和bsa摩尔比例关系的以fe2o3为无机核心的磁共振造影剂(即bsa包裹的fe2o3,以下称为fe2o3@bsa纳米颗粒),还制备了以fe3o4为无机核心的磁共振造影剂(即bsa包裹的fe3o4,以下称为fe3o4@bsa纳米颗粒),fe2o3@bsa纳米颗粒和fe3o4@bsa纳米颗粒统称为氧化铁@bsa纳米颗粒,具体操作步骤为:
54.1.1、用超纯水分别配制80mg/ml bsa溶液(稀释获得40mg/ml、20mg/ml、10mg/ml、5mg/ml、2.5mg/ml bsa溶液)、100mm naoh溶液、33mm feso4溶液和66mm fecl3溶液;
55.1.2、搭建如图1所示装置。将圆底烧瓶1置于水浴锅中,反应过程中(以下步骤1.3至步骤1.7)水浴温度固定为37℃左右(例如25℃~39℃)。用注射器吸取naoh溶液,搭载在注射泵2上。向圆底烧瓶1内通入氮气(n2)(也可以通入其他惰性气体(例如氩气等)),排尽空气。制备过程中始终保持n2通入,直至完成以下步骤1.6;
56.1.3、向圆底烧瓶1内依次加入1ml fecl3溶液和1ml feso4溶液,获得fe
2
与fe
3
的摩尔比例为1:2的混合铁盐溶液。磁力搅拌器搅拌混匀,转子转速保持在每分钟300至500转附近。此后搅拌器一直处于打开状态,直至完成以下步骤1.7;
57.1.4、按照下表1所列,向圆底烧瓶1内加入bsa溶液,搅拌混匀。此时圆底烧瓶内初始反应物状态为黄色透明溶液,将该溶液作为第一混合溶液;在优选的实施例中还可以在该第一混合溶液中加入保护试剂,例如peg及其衍生物(各种基团修饰与链长,例如peg 200、peg 400、peg 600、peg 1000、胺基peg衍生产品(例如mpeg-nhs)、巯基peg衍生产品(例如opss-peg-nh2)等)、尿素、柠檬酸、edta、以及氨基酸等,其浓度不超过50mg/ml,这类试剂的加入可以使得制备得到的氧化铁@bsa纳米颗粒尺寸更小,在体内的循环时间更长等,有利于稳态和高分辨率成像;
58.表1:bsa溶液的添加量及纳米颗粒的无机核心粒径和亲水合粒径
[0059][0060]
1.5、打开注射泵2,以12ml/h左右的注射速率向圆底烧瓶1内的第一混合溶液中缓慢逐滴滴入naoh溶液,使得第一混合溶液中naoh与fe离子总量的摩尔比例≥8:3(例如8:3~12:3,能够引起铁盐溶液中fe
2
和fe
3
全部共沉淀),naoh的加入引起fe
2
和fe
3
的共沉淀,同时使得多个bsa形成多聚体状态的蛋白纳米笼子。关闭注射泵2,此时圆底烧瓶1内溶液为黑色,ph大于10。圆底烧瓶1内溶液不超过圆底烧瓶体积的1/2;其中发生以下式(1)所示的反应:
[0061]
fe
2
2fe
3
8oh-→
fe3o4 4h2o
ꢀꢀꢀ
(1)
[0062]
1.6、继续反应1小时,此时的混合溶液作为第二混合溶液;取实验1-4的部分第二混合溶液直接按照以下步骤1.8-1.11相同的操作进行过滤、分离纯化浓缩,获得以fe3o4为核心,bsa包裹其上的纳米颗粒,命名为fe3o4@bsa纳米颗粒;
[0063]
1.7、停止向圆底烧瓶1内通入n2,向圆底烧瓶1中的第二混合溶液中滴加(例如通过注射泵2)过量(根据反应方程式计算将溶液中的fe
2
全部氧化为fe
3
的过氧化氢的基本量,加入超过基本量(例如2倍、3倍、5倍基本量等)的过氧化氢)过氧化氢溶液(双氧水,h2o2),反应1小时后结束。此时圆底烧瓶内溶液为褐红色,将此时的混合溶液作为第三混合溶液;其中发生以下式(2)所示的反应:
[0064]
2fe3o4 h2o2→
3fe2o3 h2o
ꢀꢀꢀ
(2)
[0065]
1.8、撤去反应装置,圆底烧瓶1内第三混合溶液用220nm的滤膜过滤后收集滤液;
[0066]
1.9、将所得滤液快速稀释到生理盐水或磷酸缓冲液(pbs,ph为7.2-7.4)中,生理盐水(或pbs溶液)与滤液的体积比大于10。选用截留分子量为14.4kd或30kd的透析袋透析两次,每次透析时间不少于4小时。收集透析袋中的溶液,作为透析液;
[0067]
1.10、用截留分子量为30kd的浓缩管浓缩步骤1.9得到的透析液。之后用分子筛分离浓缩后溶液中的反应产物(即fe2o3@bsa)和未反应的bsa以及其他物质。收集分子筛分离纯化的fe2o3@bsa样品峰,即得到fe2o3@bsa纳米颗粒样品溶液;
[0068]
1.11、用截留分子量为30kd的浓缩管浓缩步骤1.10得到的样品溶液,用220nm的滤膜过滤后真空管储存备用。
[0069]
通过上述步骤1.1至1.11,该实施例获得了上表1所列具有不同fe和bsa摩尔比例关系的以fe2o3为核心,bsa包裹其上的纳米颗粒,按照实验编号1-1至1-6,分别命名为fe2o3@bsa1、fe2o3@bsa2、fe2o3@bsa3、fe2o3@bsa4、fe2o3@bsa5、fe2o3@bsa6,实验1-7为在铁盐溶液中不添加bsa溶液获得的纳米颗粒,命名为fe2o3δbsa,作为对照。
[0070]
2)fe2o3@bsa纳米颗粒和fe3o4@bsa纳米颗粒的表征
[0071]
该步骤对上述步骤1)获得的纳米颗粒fe2o3@bsa1、fe2o3@bsa2、fe2o3@bsa3、fe2o3@bsa4、fe2o3@bsa5、fe2o3@bsa6、fe2o3δbsa以及fe3o4@bsa进行表征,具体操作如下。
[0072]
2.1、首先利用philips x'pert x射线粉末衍射仪上(2θ范围为10-90
°
)获取纳米颗粒的xrd图谱,并在透射电子显微镜(tem,jem 2100)上获取纳米颗粒的透射电镜图和高分辨率透射电子显微镜(hrtem),以获取尺寸和形态信息。
[0073]
2.2、随后将纳米颗粒通过尺寸排阻色谱法进行纯化,以用于负染色电子显微镜和后续实验,具体为:首先通过浓度管(30k,merck millipore,usa)将纳米颗粒分散在0.9%的nacl中,然后通过在280nm处的紫外线吸收监测将其负载在superose 6increase 10/300gl(ge healthcare)上进行洗脱纯化,确认为目的纳米颗粒的峰用醋酸铀酰进行负染色,随后进行透射电镜分析(jem 2100)。流体动力学直径(也称为亲水合粒径)(hd)通过动态光散射(英国malvern zetasizer nano zs)进行分析。在escalab220i-xl光谱仪(美国vg thermo)上检测ftir(傅立叶变换红外光谱)数据。拉曼光谱是在renishaw-invia共焦拉曼光谱仪上进行的。x射线光电子能谱(xps)数据是在escalab 250xi光电子能谱仪(thermo,usa)上获得的。实验中涉及到的金属离子(例如fe)浓度测量是在电感耦合等离子体质谱仪(icp-ms,icap qc,thermo,usa)上进行的,而bsa的浓度则是通过bca蛋白质分析试剂盒(thermo,usa)测得的。
[0074]
如上表1所示,列出了以上纳米颗粒(fe2o3@bsa1、fe2o3@bsa2、fe2o3@bsa3、fe2o3@bsa4、fe2o3@bsa5、fe2o3@bsa6、fe2o3δbsa)的核心(即无机核心)粒径以及整个纳米颗粒的粒径的检测结果,可见该实施例制备的fe2o3@bsa纳米颗粒中无机核心的粒径均小于约8.04nm,甚至可以达到3.47nm左右(fe与bsa的摩尔比为165:1,对应的fe2o3@bsa纳米颗粒亲水合粒径约为19.4nm,如图3中d幅和e幅所示,其中d幅表示fe2o3@bsa4纳米颗粒中无机核心fe2o3纳米粒子的尺寸分布,通过高斯拟合获得其平均直径为3.47
±
0.53nm,e幅表示fe2o3@bsa4纳米颗粒的亲水合粒径(hd)为19.41nm),无机核心粒径越小,最终制备得到的fe2o3@bsa纳米颗粒的亲水合粒径就越小,就越容易被吸收代谢;当不向铁盐溶液中添加bsa时,制备得到的fe2o3核心(即fe2o3δbsa)粒径高达14.50
±
3.49nm,且发生聚集堆积(如图2所示,其中a幅为tem图像,b幅为fe2o3δbsa的尺寸分布图,其中曲线采用高斯拟合获得)。如上表1所示,制备得到的fe2o3@bsa纳米颗粒的亲水合粒径均在15nm~55nm的范围内(优选当fe与bsa的摩尔比为20:1~165:1范围内时,获得的fe2o3@bsa纳米颗粒的亲水合粒径相对更小,均小于40nm;其无机核心的平均直径也相对较小),可见均具有较小的亲水合粒径。如图4中b幅所示,为fe3o4@bsa纳米颗粒中无机核心fe3o4纳米粒子的尺寸分布,通过高斯拟合获得其平均直径为3.55
±
0.65nm,与fe2o3@bsa4纳米颗粒中无机核心fe2o3纳米粒子的平均直径相当,因此其在体内也容易被吸收代谢。并且从图4中a幅也可以看出fe3o4@bsa纳米颗粒呈现出高度均一性和单分散性。
[0075]
以下以fe2o3@bsa4纳米颗粒和fe3o4@bsa纳米颗粒为例,进一步表征该实施例制备的氧化铁@bsa纳米颗粒的特性。
[0076]
如图3所示,其中a幅和b幅示出了fe2o3@bsa4纳米颗粒(即图3中所示的fe2o3@bsa,以下同)的透射电镜图,a幅分辨率为100nm,b幅为高分辨率(10nm)tem(hrtem)图像,计算的晶格间距为0.253nm(b幅右上角,分辨率2nm),与fe2o3面心立方结构的(311)面一致(如图3
中c幅所示);图3中c幅为fe2o3@bsa4和fe3o4@bsa纳米颗粒的xrd图谱,其中在2θ=23
°
处的宽峰是bsa蛋白的晶体结构域,在fe2o3@bsa4纳米颗粒的xrd图谱中311峰所在的位置代表是fe2o3晶体,而在fe3o4@bsa纳米颗粒的xrd图谱311峰所在的位置代表是fe3o4晶体;表明bsa已成功引入fe2o3@bsa4纳米颗粒或fe3o4@bsa纳米颗粒中,这与图5所示的fe2o3@bsa4、fe3o4@bsa、bsa和赤铁矿(α-fe2o3,在rruff数据库中参考r070240)的ftir光谱结果(其中特征峰的位置已用数字和垂直虚线标记)一致,其中在fe2o3@bsa4纳米颗粒的ftir光谱中,456cm-1
和595cm-1
处的峰归因于γ-fe2o3相,表明形成了γ-fe2o3。为进一步确认纳米颗粒的结构和相组成,图3中f幅示出了fe2o3@bsa4、fe3o4@bsa和赤铁矿(α-fe2o3,在rruff数据库中参考r040024)的拉曼光谱图像,其中特征峰的位置已用数字和垂直虚线标记,可见715cm-1
和1345cm-1
处的拉曼位移对应于γ-fe2o3的特征峰,表明fe2o3@bsa纳米颗粒中占主导地位的是γ-fe2o3矿物。以上结果与图6示出的xps广谱结果一致,其中在fe2o3@bsa纳米颗粒的xps广谱中,718.9ev处的卫星峰证明了fe2o3的形成,而fe3o4@bsa纳米颗粒的xps广谱在此处没有卫星峰。
[0077]
图3中g幅和h幅表示经负染的fe2o3@bsa4纳米颗粒的tem图像,其中深色中心表示存在fe2o3核,可见每个纳米颗粒均为由6~7个bsa单体围绕fe2o3核心组成的核-壳结构。为了进一步确定每个纳米颗粒的由bsa亚基组成的纳米笼结构中bsa亚基的数目,分别通过icp和bca蛋白测定试剂盒测量了纯化的fe2o3@bsa4纳米颗粒中的fe和bsa的浓度,结果如下表2所示,可见fe2o3@bsa4纳米颗粒中的fe和bsa的摩尔比大约为162.2,这与制备过程中原料fe与bsa的摩尔比165:1相当;经测量fe2o3核心的粒径约为3.5nm,对应于大约1000个fe原子,由此可以推断出每个纳米笼由大约6~7个bsa亚基构成。通过尺寸排阻色谱法(sec)进一步证实了该观察结果,如图3中i幅和图7所示,其中i幅示出了fe2o3@bsa4纳米颗粒和bsa蛋白经分子筛的洗脱曲线,图7示出了标准蛋白(购自ge healthcare)的洗脱曲线,可见fe2o3@bsa4纳米颗粒的洗脱体积对应于440kda和669kda之间的分子量,表明bsa蛋白笼的形成。同时,图8的sds-page电泳分析结果显示bsa蛋白聚集,其中泳道1为天然bsa蛋白作为对照,泳道2为经纯化后的fe2o3@bsa4峰。
[0078]
表2:fe2o3@bsa4纳米颗粒中的fe和bsa的浓度测定结果
[0079][0080]
综上可见,该实施例制备的氧化铁@bsa纳米颗粒是以fe2o3或fe3o4(也可以是两者的混合)为核心,bsa包裹其上的结构,并且由于数个bsa亚基(例如6~7个)形成的纳米笼状结构,使得在其中形成的无机核心氧化铁具有高度均一性、单分散性和粒径较小的优势,最终制备得到的氧化铁@bsa纳米颗粒的亲水合粒径也较小,易于在体内被降解代谢。
[0081]
2.3、为了测量磁性能,该实验还使用了量子设计的mpms-squid磁强计(xp-5xl型),其中以fe2o3@bsa4纳米颗粒和fe3o4@bsa纳米颗粒(实施例1制备)为例,使用纳米颗粒的冻干粉作为样品,并以gd-dtpa(magnevist)作为对照。在300k下测量了-2t~2t的磁化曲线,在100oe下测量了10k~300k的零场冷却(zfc)和场冷(fc)曲线。
[0082]
2.4、为了检验使用fe2o3@bsa纳米颗粒和fe3o4@bsa纳米颗粒作为mri造影剂的可
行性,该实验还在3t临床mri扫描仪(achievea,飞利浦医疗系统公司,best,荷兰)上测量了fe2o3@bsa4纳米颗粒和fe3o4@bsa纳米颗粒的弛豫时间。具体操作为:将1.5ml具有不同fe元素浓度(铁元素浓度分别为0.8、0.4、0.2、0.1、0.05mm)的fe2o3@bsa4纳米颗粒、fe3o4@bsa纳米颗粒或gd-dtpa的去离子水溶液转移到2ml离心管中,利用3t临床mri扫描仪进行测量。其中通过拟合1/t1和1/t2值与铁浓度的关系来计算弛豫值。采用涡轮自旋回波(tse)序列获得t1加权磁共振图像,其参数如下:tr/te=150、300、600、1000、2000、4000、8000/11ms,层厚3mm,翻转角=90
°
,平均信号数为2个,视野(fov)=120mm
×
120mm,矩阵尺寸=240
×
240。采用多自旋回波(mse)序列获得t2加权mr图像,除tr/te=5000/10、20、30、40、50、60、70、80、90、100ms外,其余参数与t1均相同。
[0083]
如图9所示,示出了fe2o3@bsa4纳米颗粒与gd-dtpa的磁性能对比结果,其中a幅为在300k时与磁场有关的磁化曲线(m-h)和矫顽力(hc)(放大插图),可见,fe2o3@bsa4纳米颗粒在300k时的饱和磁化强度(m)仅为55emu/g fe(显著低于超顺磁性纳米颗粒如ferumoxytol(nat.biomed.eng.2017,1(8),637-643,doi:10.1038/s41551-017-0116-7.)在300k时的饱和磁化强度95emu/g fe),并且与典型的超顺磁性纳米颗粒如ferumoxytol相比,由于fe2o3@bsa4纳米颗粒的体积各向异性很小,其矫顽力和顽磁在室温下可以忽略不计。图9中b幅为在100oe下零场冷却(zfc)和场冷却(fc)后的温度相关磁化曲线(m-t),可见测得的fe2o3@bsa4纳米颗粒的阻断温度(tb)仅为23k,远小于典型的超顺磁性纳米颗粒如ferumoxytol的53k,以上表明fe2o3@bsa4纳米颗粒的粒径相对更小;如图10所示,其中a幅示出了fe3o4@bsa纳米颗粒在300k时与磁场有关的磁化曲线(m-h),b幅为fe3o4@bsa纳米颗粒在100oe下零场冷却(zfc)和场冷却(fc)后的温度相关磁化曲线(m-t),可见fe3o4@bsa纳米颗粒在300k时的饱和磁化强度(m)为约65emu/g fe,略高于fe2o3@bsa4纳米颗粒,同样显著低于超顺磁性纳米颗粒如ferumoxytol;测得的fe3o4@bsa纳米颗粒的阻断温度(tb)约为25k,与fe2o3@bsa4纳米颗粒相当,也远小于典型的超顺磁性纳米颗粒如ferumoxytol,表明fe3o4@bsa纳米颗粒的粒径也较小,因此相对于典型的超顺磁性纳米颗粒如ferumoxytol,本发明提供的氧化铁@bsa纳米颗粒更有利于作为磁共振造影剂(优选为t1造影剂)。
[0084]
图9中c幅和d幅分别为fe2o3@bsa4纳米颗粒的t1弛豫率图和t2弛豫率图,可见fe2o3@bsa4纳米颗粒的r1为6.81mm-1
·
s-1
,r2值为72.37mm-1
·
s-1
(r2/r1值为10.6),gd-dtpa的r1值3.88mm-1
·
s-1
,r2值为7.18mm-1
·
s-1
;图11中a幅为fe3o4@bsa纳米颗粒的t1和t2弛豫率图,b幅为fe3o4@bsa纳米颗粒的t1和t2加权mr图像,可见fe3o4@bsa纳米颗粒的r1为6.41mm-1
·
s-1
,r2值为86.24mm-1
·
s-1
(r2/r1值13.5)。可见fe3o4@bsa纳米颗粒和fe2o3@bsa4纳米颗粒的r2/r1值相差不大,两者的r1值也相当,且均远大于gd-dtpa的r1值,r1值的增加和/或r2/r1值的降低都有助于增强作为t1造影剂的对比效果。图9中e幅和f幅分别示出了t1加权mr图像和t2加权mr图像,即fe2o3@bsa4纳米颗粒和gd-dtpa的水溶液mri成像结果的实物图。以上结果表明该实施例制备的氧化铁@bsa纳米颗粒具有作为磁共振造影剂的潜力,并证明了该实施例制备的氧化铁@bsa纳米颗粒作为磁共振造影剂以测试mri的适用性,且在临床强度施加的磁场下显示出较低的磁化强度,更有利于作为t1造影剂。
[0085]
实施例2:磁共振成像纳米造影剂的制备
[0086]
该实施例旨在利用多种具有不同fe
2
和fe
3
摩尔比的铁盐溶液来制备磁共振成像纳米造影剂,其与实施例1提供的制备方法的不同之处仅在于:按照下表3所列制备铁盐溶
液(其中以feso4溶液和fecl3溶液的添加总量为2ml为例),随后均按照fe与bsa的摩尔比为165:1添加bsa溶液,然后添加naoh溶液(按照实施例1提供的制备方法中步骤1.1至1.6)或依次加入naoh溶液和过量的过氧化氢溶液(按照实施例1提供的制备方法中步骤1.1至1.11),最终制备得到一系列氧化铁@bsa纳米颗粒。
[0087]
表3:具有不同fe
2
和fe
3
摩尔比的铁盐溶液
[0088][0089]
经对上表3所列实验2-1至2-8制备得到的氧化铁@bsa纳米颗粒按照上述实施例1中2.1至2.4的方法进行检测,所有氧化铁@bsa纳米颗粒均为以氧化铁(fe2o3、fe3o4或两者混合)为核心,bsa包裹于该核心氧化铁上的结构,且其无机核心氧化铁粒径均在3.5nm左右(3.0nm~5.0nm),氧化铁@bsa纳米颗粒的亲水合粒径均在20nm左右(15nm~35nm),1.5t到3t磁场范围内的r1值为4.5-18mm-1
s-1
,1.5t到3t磁场范围内的r2/r1的比值为5到16.5,均可作为磁共振造影剂,且更有利于作为t1造影剂。
[0090]
如上表3所示,在不同的fe
2
和fe
3
摩尔比的铁盐溶液中加入的naoh溶液的量只要满足使得铁盐溶液中的fe
2
和fe
3
能够发生共沉淀即可,因此加入的naoh溶液的量可以为使得溶液中的naoh与fe离子总量的摩尔比为≥2:1(例如2.2:1~2.8:1,可以使得上表3所列铁盐溶液中的fe
2
和fe
3
全部共沉淀或者部分共沉淀),优选为≥3:1,该摩尔比可以使得任意fe
2
和fe
3
摩尔比的铁盐溶液中的fe
2
和fe
3
全部共沉淀。当需要加入h2o2以制备以fe2o3为核心的fe2o3@bsa纳米颗粒时,h2o2的加入量只要满足使溶液中所有的fe
2
均转化为fe
3
即可,但是在实验中往往加入更多量的h2o2,保证fe
2
的氧化转化更彻底。
[0091]
还可以利用与实施例1和实施例2相同的方法制备并表征以氧化铁为核心,人血清白蛋白(has)、人铁蛋白、dna结合蛋白或热休克蛋白等功能化蛋白中的一种或多种的混合包裹其上的纳米颗粒,其也具有作为磁共振造影剂(例如t1或t2造影剂)的潜力。
[0092]
实施例3:fe2o3@bsa纳米颗粒的体内mr造影
[0093]
该实施例以上述实施例1制备的fe2o3@bsa4纳米颗粒为例以对本发明提供的纳米颗粒进行体内mr造影分析,具体包括以下操作。
[0094]
3.1、通过尾静脉将fe2o3@bsa4纳米颗粒或gd-dtpa(magnevist)(每千克大鼠体重0.15mmol fe/gd)静脉注射到6周龄的雄性sprague dawley(sd)大鼠(购自南模生物,平均体重200g,所有大鼠均按照《实验室动物护理和使用指南》中概述的指南接受护理)中。
[0095]
3.2、腹腔注射5%水合氯醛麻醉大鼠,检查注射前和注射后2分钟、5分钟、15分钟、
30分钟、45分钟、60分钟的矢状切片(即mri扫描大鼠结果)。mr造影图像采用3t临床mri扫描仪获得,其中采用三维快速场回波(ffe)序列获取t1加权图像,fov=18cm
×
18cm
×
8.4cm,矩阵=360
×
256
×
140,分辨率=0.5mm
×
0.7mm
×
0.6mm。tr=25ms,te=3.5ms,翻转角=30
°
,平均数=1。
[0096]
如图12所示,其中a幅表示在3t下注射fe2o3@bsa4纳米颗粒和gd-dtpa后,大鼠的t1加权mr图像的比较。每个图像上方的时间点是尾静脉注射后的时间。pre表示注射之前。可见在注射fe2o3@bsa4纳米颗粒之前,心脏(箭头h)和肾脏(箭头k)没有显示出明显的正对比。注射后,心脏和肾脏在注射后立即显示出较高的正对比度。t1信号在注射后15-30分钟持续增强,直到达到最亮,这将大大有助于病理血管组织的检测。然后直到注射后60min,t1信号逐渐消失。图12中b幅表示注射后心脏的信噪比(snr)的对比度增强比,其中snr变化(%)=100%
×
(snr
post-snr
pre
)/snr
pre
;图13示出了注射前和注射后心脏的信噪比(snr);通过比较血管系统的信噪比(snr),可以进一步定量评价fe2o3@bsa4纳米颗粒作为造影剂的应用。从图13和图12中b幅所示结果,可见心脏snr从注射前的45.2上升到注射后15分钟的78.1,然后逐渐下降到注射后60分钟的50.5,在注射后15分钟,信噪比增加了近73%。图12中c幅表示在3t下注射fe2o3@bsa4纳米颗粒的大鼠的t1加权mr血管造影(mra)的3d图像。每个图像下方的时间点是注射后的时间。可见注射fe2o3@bsa4纳米颗粒10分钟后,mra显示出心脏(箭头h)和颈总动脉(箭头ca)的强烈正对比,而随着时间的流逝逐渐消失,表明fe2o3@bsa4纳米颗粒可以被代谢。
[0097]
由图12中a幅可见,以注射相同剂量的gd-dtpa作为对照,注射2min后可立即观察到具有高对比度效果的血池图像。但t1信号在注射后5min迅速消失,在注射后30min时几乎消失。另外,在注射gd-dtpa 2分钟后,大鼠的组织间隙也出现了信号强度增强,全身出现了亮信号。由图13和图12中b幅可见,注射gd-dtpa后,心脏snr由注射前的44.5增加到注射后2分钟的74.3,然后急剧下降到注射后30分钟的52.7,因此在注射gd-dtpa后2分钟,信噪比增加了近67%,具有较强的增加,但是相对于本发明提供的fe2o3@bsa4纳米颗粒持续时间较短,不利于临床上稳定造影。虽然重复注射gd-dtpa也可以连续获得高分辨率mra,但在临床应用中可能会增加gd沉积的风险及在体内的相关有害影响。
[0098]
综上实施例结果表明,相对于临床使用的gd-dtpa造影剂的造影效果,本发明提供的fe2o3@bsa纳米颗粒作为造影剂时的信号更强,其血液半衰期更长,可以延长血管造影效果,使其能够获得介入手术或者其他非医学目的造影所需的稳态成像和高分辨率图像,并可以帮助减少心脏和血管边界的模糊,提高图像的稳定性和清晰度,这表明本发明提供的fe2o3@bsa纳米颗粒在作为磁共振造影剂方面具有潜在的高效率和高稳定性。
[0099]
按照与以上实施例3相同的方法也验证了实施例1中制备的fe3o4@bsa纳米颗粒的体内mr造影效果,其也获得了类似于fe2o3@bsa4纳米颗粒的造影效果,表明本发明提供的氧化铁@bsa纳米颗粒在作为磁共振造影剂(例如t1造影剂)方面具有潜在的高效率和高稳定性。
[0100]
实施例4:fe2o3@bsa纳米颗粒的体外和体内毒性分析和稳定性评估
[0101]
该实施例以上述实施例1制备的fe2o3@bsa4纳米颗粒为例以对本发明提供的纳米颗粒进行体外和体内毒性分析和稳定性评估,具体包括以下操作。
[0102]
4.1、体外稳定性评价:首先将fe2o3@bsa4纳米颗粒分散在pbs中(ph=7.2),在真空
管中室温储存14天,储存期间通过cd和dls(动态光散射)测量评价其时间依赖性稳定性。铁浓度依赖的hd通过dls测量进行了仔细的监测。对于ph依赖性稳定性评估,将fe2o3@bsa4纳米颗粒在不同ph(6.0~8.5)的pbs中室温孵育24h,孵育后测定紫外-可见吸收和hd。
[0103]
结果如图14和图15所示,其中图14中a幅表示以bsa为对照,fe2o3@bsa4纳米颗粒在pbs中储存不同时间的cd光谱,可见受载bsa的二次结构受铁芯的影响不大,与原生bsa结构基本一致;b幅示出了fe2o3@bsa4纳米颗粒的时间依赖性hd照片,可见在室温下孵育14天后,没有明显的沉淀,hd几乎恒定;c幅示出了fe2o3@bsa4纳米颗粒在不同fe浓度(fe浓度为25、50、125、250、500和1000μg/ml)下的hd,可见即使在fe浓度达到1000μg/ml时,也没有明显的hd变化和可见的沉淀。图15示出了fe2o3@bsa4纳米颗粒在不同ph(6.0~8.5)下的hd变化,可见fe2o3@bsa4纳米颗粒在不同ph值下均无明显变化。纳米颗粒的稳定性主要归因于其周围的bsa壳层,赋予了纳米颗粒良好的水溶性和缓冲能力,而且bsa涂层的表面电荷排斥可能会阻碍纳米颗粒的聚集。这些结果表明,本发明提供的fe2o3@bsa纳米颗粒具有良好的体外稳定性,适合进一步的生物医学应用。
[0104]
4.2、安全性评价:为了评估fe2o3@bsa纳米颗粒的安全性,该实验使用hepg2和293t细胞系(购于中国科学院),通过活/死染色和cck-8检测暴露于不同fe浓度的纳米颗粒的hepg2和293t细胞测定fe2o3@bsa4纳米颗粒的细胞毒性,具体操作为:将hepg2细胞和293t细胞接种于密度为5000个/孔的96孔板中培养12h,然后在含有fe2o3@bsa4纳米颗粒(铁浓度:0、10、25、50、100和300μg/ml)的条件下持续培养24小时。随后,收集细胞,用钙黄绿素am(2μm)和pi(4μm)染色液离光培养15~20min,最后在荧光显微镜下拍摄细胞荧光图像。细胞活力测定采用计数试剂盒-8(cck-8),将hepg2细胞和293t细胞在含有fe2o3@bsa4纳米颗粒(铁浓度:0、10、25、50、100和300μg/ml)条件下培养24小时,然后用10%(v/v)cck-8溶液孵育2小时,通过读取450nm处的吸光度计算每个样品的细胞存活率。每个测试有三个平行的样本。
[0105]
图14中d幅示出了hepg2和293t细胞的cck-8细胞活力测定结果,可见各组中几乎没有死细胞,即使在fe浓度达到300μg/ml时,也几乎没有死细胞,所有实验组的细胞存活率都大于95%。
[0106]
4.3、溶血评价:以评估血液相容性,还进行了溶血试验,具体操作为:用新鲜大鼠血液制备红细胞悬液(rbc,2%)。溶血试验用500μl红细胞悬液混合fe2o3@bsa4纳米颗粒获得不同铁浓度(0、10、25、50、100和300μg/ml)的溶液作为样品,去离子水和pbs分别作为阳性对照和阴性对照。在37℃水浴中孵育1h后,以13000rpm离心5min。收集上清液以测量570nm处的光密度(od)。溶血率按下式计算,溶血率(%)=(od
样品
–
od
阴性
)/(od
阳性
–
od
阴性
)
×
100%。一般认为当溶血率低于5%时不会发生溶血。
[0107]
结果如图14中e幅所示,可见即使在fe浓度高达300μg/ml的纳米颗粒浓度下也没有检测到溶血,表明本发明提供的fe2o3@bsa纳米颗粒与血细胞高度相容。
[0108]
4.4、时间依赖性的生物分布:通过尾静脉将fe2o3@bsa4纳米颗粒的生理盐水溶液(每千克大鼠体重0.15mmol fe)注射到大鼠体内。对照组为生理盐水溶液。分别于注射后24h和48h处死大鼠,收集心、肝、脾、肺、肾等脏器。精确称量大鼠的身体和器官重量,然后从每个器官的不同部位切下三小块组织转移到2ml离心管中称重并计算每个组织的湿重。酸消化后,用icp-ms法测定各器官中铁元素的含量。
[0109]
为评价fe2o3@bsa纳米颗粒体内的长期组织毒性,分别在注射后7天和14天进行h&e组织病理学检查。以注射生理盐水的大鼠为对照。所有器官(心、肝、脾、肺、肾等脏器)标本用4%多聚甲醛固定,固定后的器官进行石蜡包埋苏木精-伊红(h&e)组织病理学检查。
[0110]
研究肾清除率时,将fe2o3@bsa4纳米颗粒(每千克大鼠体重0.15mmol fe)尾静脉注射大鼠,分别于注射前、注射后2h、4h、6h、12h、24h、48h采集尿样。用icp-ms(美国thermo公司)测量每个样品的铁含量,用tem(jem 2100)观察尿液中的纳米颗粒。
[0111]
还分别于注射后7天和14天采集大鼠血清,进行血液生化分析。
[0112]
如图14中f幅所示,为尾静脉注射后24h和48h后主要器官中fe元素的时间依赖性生物分布分析,可见在注射后24h,fe2o3@bsa4纳米颗粒主要集中在肝脏、脾脏和肾脏中。结果与上面的mr造影成像高度一致,并且大多数纳米颗粒可以在注射后48h内从体内有效清除。
[0113]
如图16所示,为注射7天和14天后收获的大鼠主要器官的h&e染色图像(比例尺为100μm)。可见,与对照组相比,注射fe2o3@bsa4纳米颗粒的大鼠主要器官没有表现出任何明显的异常。表明该纳米颗粒具有良好的体内生物相容性,这种高生物相容性主要是由于fe2o3@bsa4纳米颗粒中fe2o3与bsa的结合。
[0114]
如图17所示,为注射前、注射后2h、4h、6h、12h、24h、48h大鼠尿液中的fe含量,可见注射后2h,检测到尿液中铁含量最高,在注射后12h,尿液中的铁含量下降80%,并在注射后48h恢复正常。在注射后2h和24h分别采集尿液标本进行tem检测,如图18中a幅和b幅所示,可见注射后2h尿液中可观察到大量的fe2o3@bsa4纳米颗粒(a幅),而注射后24h尿液中几乎检测不到fe2o3@bsa4纳米颗粒(b幅)。显然,fe2o3@bsa4纳米颗粒可以通过肾脏迅速清除。
[0115]
如图18中c幅所示,为注射fe2o3@bsa4纳米颗粒后,各血液生化指标的变化情况,其中alb:白蛋白;alp:碱性磷酸酶;alt:丙氨酸转氨酶;ast:天冬氨酸转氨酶;bun:血尿素氮;cre:肌酐;tbil:总胆红素;ldh:乳酸脱氢酶。可见对照组和实验组之间无明显差异,说明该纳米颗粒注射后未出现肾损伤和肾清除引起的明显副作用。
[0116]
按照与以上实施例4相同的方法也对实施例1制备的fe3o4@bsa纳米颗粒进行了体外和大鼠体内的毒性分析和稳定性评估,其也获得了类似于fe2o3@bsa纳米颗粒的效果。表明本发明提供的氧化铁@bsa纳米颗粒具有较高的生物相容性和生物安全性,在体内容易被降解和被肾脏代谢清除,潜在的肾脏毒性较小,可以作为一种低肾毒性蛋白-氧化铁复合纳米磁共振造影剂。
[0117]
按照与以上相同的方法,还证明了本发明提供的氧化铁@bsa纳米颗粒在家兔、比格犬体内具有与在大鼠体内相似的血管造影性能和安全性评价结果。
[0118]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。