一类urat1抑制剂及其制备方法与用途
技术领域
1.本发明具体涉及一类urat1抑制剂及其制备方法与在高尿酸血症和痛风治疗上的用途。
背景技术:
2.尿酸是内源性和膳食嘌呤代谢的最终产物,尿酸在血液中作为抗氧化剂存在,由于人体内缺乏降解尿酸的尿酸酶,尿酸主要通过肾脏和肠道排出体外,其中肾脏是尿酸排泄的主要途径,余下经粪便排泄。尿酸生成过多、排泄不足或两者并存的情况都会导致高尿酸血症,高尿酸血症是指血液中的尿酸浓度超出正常范围一种机体状态,一般男性超过417μmol/l,女性超过357μmol/l,研究表明90%是因为尿酸排泄减少所致。近年来,随着人们生活水平的提高,高尿酸血症患病率有明显上升,2009年上海地区高尿酸血症患病率为10.0%;北京市1120例受试者高尿酸血症患病率为17.86%;广州地区患病率居全国首位,男性27.9%,女性12.4%,总患病率高达21.81%。
3.高尿酸血症最终可能会导致炎症、变形结节形成、严重疼痛间歇发作和肾脏疾病,所以过去常把高尿酸血症与痛风和肾脏疾病的发病相联系,认为高尿酸血症是痛风及肾脏疾病发病过程中的一个阶段。现代研究表明,高尿酸血症不仅与痛风和肾脏疾病相关,还与心血管疾病、糖尿病、代谢综合征、高脂血症等疾病的发生发展关系密切,已经成为这些疾病的独立危险因素。目前,高尿酸血症、痛风和糖尿病一样已成为严重威胁人类健康的代谢性疾病,联合国将其列为21世纪20大顽症之一。
4.降低血尿酸水平的药物包括:抑制尿酸生成的酶抑制剂,如别嘌呤醇、非布司他和硫嘌呤醇等黄嘌呤氧化酶抑制剂;促进尿液中尿酸排泄的药物,如丙磺舒、苯溴马隆和lesinuard等;迅速降低血尿酸的尿酸分解剂,如pegloticase等聚乙二醇重组尿酸酶;用于痛风急性发作控制的消炎止痛药物,如秋水仙碱和非甾体抗炎药物(nsaid)等。据统计,治疗高尿酸血症/痛风药物主要分布在前两者,分别为抑制尿酸生成的黄嘌呤氧化酶xo抑制剂和尿酸转运蛋白urat1抑制剂。其中,有关别嘌醇的严重药物超敏反应和非布司他的心脏毒性风险对黄嘌呤氧化酶xo抑制剂发展产生了巨大不利影响。从靶点安全性或产品临床试验药效出发,urat1抑制剂成为目前该领域新药开发唯一突破点,尿酸转运体1(urate transporter 1,urat1)是重要尿酸转运体,尿酸在近端肾小管的重吸收大部分依靠urat1完成,urat1不受膜电压和细胞内外ph值的影响,抑制urat1就会抑制肾脏中尿酸的重吸收、增加尿液中尿酸的排泄,进而达到降低血尿酸和控制痛风发作的目的。相较于别嘌醇和非布司他等黄嘌呤氧化酶抑制剂,urat1抑制剂的作用机制更加合理,因为只有10%患者存在尿酸生成过多,其余90%患者均有不同程度的肾脏排泄不足。但目前上市药物都存在各自风险,其中,丙磺舒能引发肾绞痛和肾功能损害;苯溴马隆发生过肝脏损伤甚至肝衰竭,在一些国家已经陆续撤市;lesinuard被fda发布肾衰黑框警告,已于2019年退市。
5.综上所述,现有药物相关不可忽略的不良反应逐渐超过现有治疗益处,为了满足未来临床需求,迫切需要研发新型高效、低毒urat1抑制剂。
技术实现要素:
6.本发明提供了一种新型结构urat1抑制剂,并发现具有此类结构的化合物具有良好的活性,并表现出优异的降血清尿酸浓度,治疗高尿酸血症和痛风的作用。
7.本发明的目的可以通过以下方案达到:
8.一类式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其药学上可接受的盐:
[0009][0010]
其中:
[0011]
w1各自独立选自n或cra;
[0012]
ra为h或h被一个或多个选自卤素、氰基、硝基、氨基、羟基、氧代基、c
1-6
烷基、c
1-6
卤代烷基、c
1-6
羟烷基、c
1-6
烷氧基的取代基所取代;
[0013]
r1选自c
1-6
烷基、c
1-6
取代烷基、c
3-6
环烷基、c
3-6
取代环烷基、c
3-6
杂环基、c
3-6
取代杂环基、c
6-10
芳基、c
6-10
取代芳基、c
6-10
杂芳基或c
6-10
取代杂芳基,其中所述的c
1-6
取代烷基、c
3-6
取代环烷基、c
3-6
取代杂环基、c
6-10
取代芳基或c
6-10
取代杂芳基上的h分别独立地任选进一步被一个或多个选自卤素、羟基、氰基、硝基、醚基、酯基、氨基、酰胺基、c
1-6
烷基、c
1-6
卤代烷基、c
6-10
芳基的取代基所取代;
[0014]
r2、r3分别独立地选自h、卤素或c
1-6
烷基,或者r2和r3及连接r2和r3的碳共同构成3-6元环烷基,r6和r7及连接r6和r7的碳共同构成3-6元环烷基;
[0015]
r4为h、c
1-6
烷基或取代的c
1-6
烷基,其取代基选自c
1-2
烷氧基、羟基或氨基。
[0016]
进一步,优选的,所述的式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其可药用盐,其中所述的ra为氢原子、卤素、氰基、硝基、氨基、羟基、氧代基、c
1-6
烷基、c
1-6
卤代烷基,优选为氢原子、卤素、氰基、c
1-6
卤代烷基。
[0017]
进一步,优选的,所述的式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其可药用盐,其中所述的r1选自c
1-6
烷基、c
1-6
取代烷基、c
6-10
芳基、c
6-10
取代芳基、c
6-10
杂芳基、c
6-10
取代杂芳基,所述的c
1-6
取代烷基、c
3-6
取代杂环基、c
6-10
取代芳基或c
6-10
取代杂芳基上的h分别独立地任选进一步被一个或多个选自羟基、醚基、c
1-6
烷基、c
1-6
卤代烷基、苯基的取代基所取代;再进一步,所述的r1各自独立选自c
1-6
羟烷基、吡啶基、喹啉基或被一个或多个羟基、c
1~6
的烷基、卤素、醚基、取代的烷基、苯基、吡啶基或喹啉基;更进一步,所述的r1各自独立选自c
2~4
羟烷基、被苯环取代的羟甲基、吡啶基、喹啉基或吡啶上的h被甲基、甲醚基、f、cl单取代或多取代。
[0018]
进一步,优选的,所述的式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其可药用盐,其中所述的r2、r3分别独立地选自c
1-6
烷基,或者r2和r3及连接r2和r3的碳共同构成4元环烷基;再进一步,所述的r2、r3分别独立地选自甲烷,或者r2和r3及连接r2和r3的碳共同构成4元环烷基。
[0019]
进一步,优选的,所述的式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其药学上可接受的盐,其中所述的r4或r8选自h、c
1-6
烷基或c
1-6
取代烷基,再进一步所述的r4选自h。
[0020]
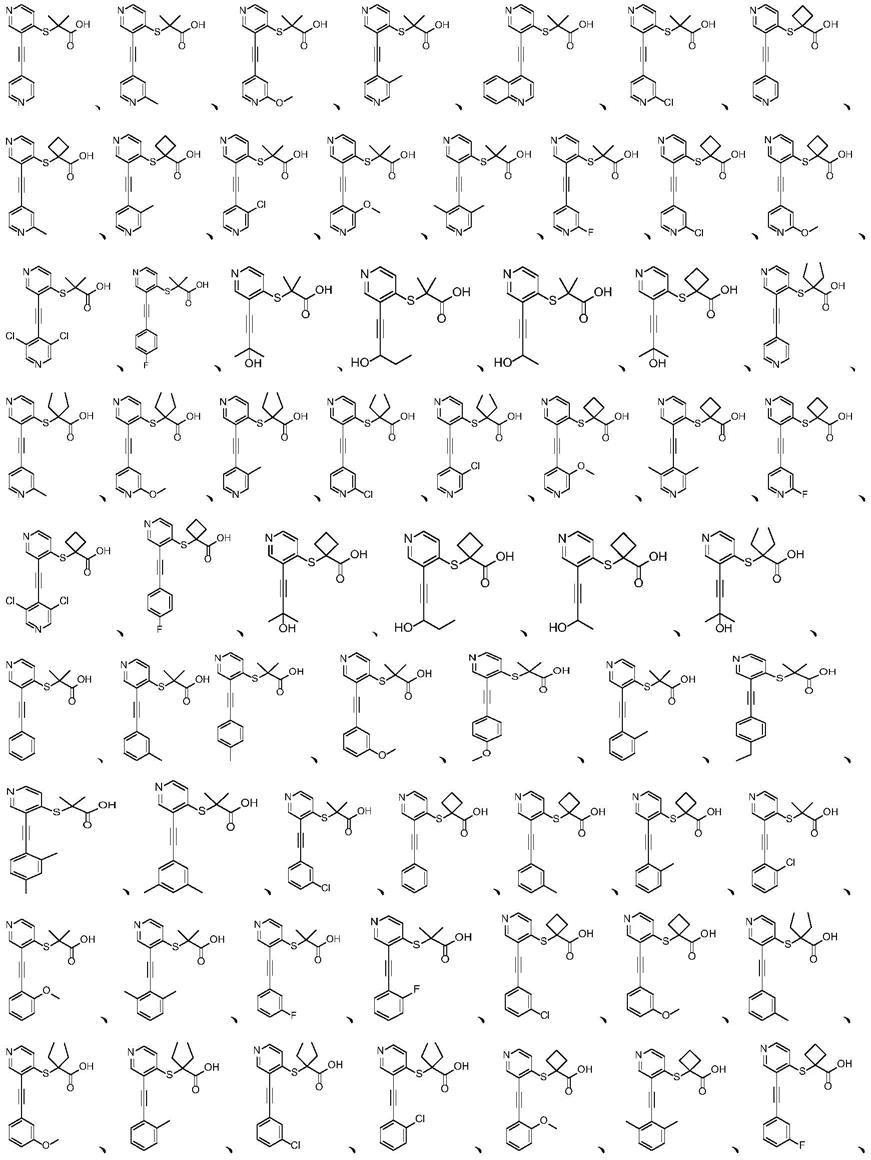
进一步,所述的式i所示的化合物选自下列之一:
[0021]
[0022][0023]
进一步,本发明所述的式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其可药用盐的制备方法包括:
[0024][0025]
通式i-a化合物与通式i-b化合物在碱性条件下进行反应,得到通式i-c化合物,在金属催化剂下与通式i-d化合物进行偶联反应,然后在碱性条件下水解反应得到通式i化合物;其中:x1和z1为卤素,所述卤素优选自氯、溴、碘;y1选自氢原子或钠原子;所述无机碱优选自碳酸钾、碳酸钠、碳酸铯、氢化钠、氢氧化钠、氢氧化钾;所述有机碱优选自三乙胺、二异丙基乙胺;所述的r1~r4的定义如上所述。
[0026]
进一步,所述的式i-a化合物与式i-b化合物的物质的量之比为1:0.2~1:5,式i-c化合物与式i-d化合物及金属催化剂的物质的量之比为1:0.2:0.01~1:5:1。
[0027]
进一步,所述反应所用溶液为非质子性溶剂,选自四氢呋喃、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、乙腈。
[0028]
进一步,所述的碱性物质选自无机碱或有机碱,所述无机碱优选自碳酸钾、碳酸钠、碳酸铯、氢化钠、氢氧化钠、氢氧化钾;所述有机碱优选自三乙胺、二异丙基乙胺。所述的碱性物质的加入量以所述的式i-a化合物的物质的量计为0.2~5mol/mol。
[0029]
进一步,所述的金属催化剂选自过渡金属钯、金属铜、铁、金中至少一种,优选自过渡金属钯、铜或者两者的混合。
[0030]
本发明所述的药物组合物,所述药物组合物含有治疗有效量的所述的通式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式,及其可药用盐,以及一种或多种药学上可接受的辅料。
[0031]
进一步,本发明所述的药物组合物含有另外一种或多种降尿酸药物,所述降尿酸
药物选自urat1抑制剂、黄嘌呤氧化酶抑制剂、黄嘌呤脱氢酶或黄嘌呤氧化还原酶抑制剂。
[0032]
更进一步,本发明所述的药物组合物,其进一步含有另外一种或多种降尿酸药物,所述降尿酸药物选自别嘌呤醇、非布司他或托匹司他。
[0033]
本发明所述的通式i所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体及其混合物形式及其可药用盐或所述的药物组合物在制备抑制urat1的药物中的用途。
[0034]
进一步,所述抑制urat1的药物是预防和/或治疗痛风、复发性痛风发作、痛风性关节炎、高尿酸血症、高血压、心血管疾病、冠心病、莱-萘二氏综合症、凯-赛二氏综合症、肾病、肾结石、肾衰竭、关节炎症、关节炎、尿石症、铅中毒、甲状旁腺功能亢进、银屑病、结节病或次黄嘌呤-鸟嘌呤磷酸核糖转移酶缺乏症的药物,优选预防和/或治疗痛风或高尿酸血症的药物。
[0035]
与现有技术相比,本发明的有益效果在于:
[0036]
本发明公开了一类urat1抑制剂及其制备方法与在制备抑制urat1的药物中的用途,本发明提供的各种化合物及其盐类、水合物或溶剂合物,是一种选择性尿酸再吸收抑制剂,可以通过促进尿酸从体内排泄并减少血清尿酸来治疗高尿酸血症和痛风,对尿酸转运蛋白1具有很好的抑制效果,且在动物体内表现出高效降低尿酸且低毒性的效果。
具体实施方式
[0037]
给出下列制备实施例和生物学实施例,使本领域技术人员能够更清楚地理解和实施本发明。它们不应被解释为限制本发明的范围,仅仅是其例证和代表。
[0038]
反应式如下:
[0039][0040]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0041]
化合物的结构由核磁共振(nmr)或质谱(ms)来确定,核磁共振谱是通过bruker avance-500仪器获得,氘代二甲亚砜,氘代氯仿和氘代甲醇等为溶剂,四甲基硅烷(tms)为内标。质谱是由液相色谱-质谱(lc-ms)联用仪agilent technologies 6110获得,采用esi离子源。
[0042]
实施例1
[0043]
中间体a1的合成
[0044][0045]
向50ml的反应瓶中加入4-氯-3碘吡啶(500mg,2.09mmol),硫化钠(195.46mg,2.50mmol),dmf(3ml),反应液在80℃下搅拌4h。反应完毕后,加入20ml水,用1mol
·
l-1
的稀盐酸调节ph至5~6,析出淡黄色固体,抽滤得到中间体a1(405mg),收率82%。
[0046]
中间体b1的合成
[0047][0048]
向50ml的反应瓶中加入中间体a(400mg,1.69mmol),2-溴异丁酸乙酯(337.62mg,2.03mmol),碳酸铯(1.66g,5.08mmol),dmf(5ml),反应液在60℃下搅拌4h。反应完毕后,加入20ml水,用乙酸乙酯萃取三次(3
×
50ml),有机层合并,饱和食盐水洗,无水硫酸钠干燥,过滤,旋蒸后过柱得中间体b1(410mg),收率69%。
[0049]
中间体c1的合成
[0050][0051]
向50ml反应瓶中加入4-溴吡啶(316.00mg,2.00mmol),三甲基硅基乙炔(235.20mg,2.40mmol),pd(pph3)2cl2(70.19mg,0.1mmol),cui(38.09mg,0.20mmol),et3n(607.14mg,6.00mmol),乙腈5ml,氮气置换3次后,氮气保护下80℃反应4h。反应完毕后,反应液硅藻土助滤,乙酸乙酯洗涤滤饼,滤液旋蒸后过柱得中间体c1(208mg),收率58%。
[0052]
中间体d1的合成
[0053][0054]
向50ml反应瓶中加入中间体b1(386.32mg,1.10mmol),中间体c1(175.31mg,1.00mmol),pd(pph3)2cl2(175.31mg,0.25mmol),ag2o(463.48mg,2.00mmol),dmf(5ml),氮气置换3次后,氮气保护下70℃反应8h。反应完毕后,反应液硅藻土助滤,甲醇洗涤滤饼,滤液旋蒸后过柱得中间体d1(175mg),收率50%。
[0055]
2-((3-(吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸e1(化合物1)合成
[0056]
[0057]
向50ml的反应瓶中加入中间体d1(163.21mg,0.50mmol),用2mol
·
l-1
lioh水溶液(2.50ml,5.00mmol),甲醇(10ml),室温反应5h。反应完毕后,反应液旋干,加入10ml水溶解后过滤,滤液用1n盐酸中和至ph为5-6,析出固体,过滤,滤饼烘干得化合物1(97mg),收率67%。1h nmr(500mhz,dmso-d6)δ13.27(s,1h),8.79-8.59(m,3h),8.50(d,j=5.5hz,1h),7.60-7.48(m,2h),7.39(d,j=5.5hz,1h),1.63(s,6h).ms:298.75(m
).
[0058]
实施例2
[0059]
2-((3-(2-甲基-吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物2)合成,将4-溴-2-甲基吡啶(344.06mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物2(72mg),总收率14%,化合物2结构如下:
[0060][0061]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.60(s,1h),8.53(d,j=5.1hz,1h),8.40(d,j=5.5hz,1h),7.51(d,j=5.5hz,1h),7.42(s,1h),7.34(dd,j=5.1,1.6hz,1h),1.58(s,9h).ms:312.82(m
).
[0062]
实施例3
[0063]
2-((3-(2-甲氧基-吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物3)合成,将4-溴-2-甲氧基吡啶(376.04mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物3(79mg),总收率15%,化合物3结构如下:
[0064][0065]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.67(s,1h),8.47(d,j=5.5hz,1h),8.29-8.20(m,1h),7.41(d,j=5.5hz,1h),7.13(dd,j=5.3,1.3hz,1h),6.98(t,j=1.1hz,1h),3.89(s,3h),1.61(s,6h).ms:328.83(m
).
[0066]
实施例4
[0067]
2-((3-(3-甲基-吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物4)合成,将4-溴-3-甲基吡啶(344.06mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物4(40mg),总收率8%,化合物4结构如下:
[0068][0069]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.71(s,1h),8.59(s,1h),8.49(d,j=5.2hz,2h),7.49(d,j=5.0hz,1h),7.42(d,j=5.5hz,1h),1.61(s,9h).ms:312.57(m
).
[0070]
实施例5
[0071]
2-((3-(喹啉-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物5)合成,将4-溴喹啉(416.12mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物5(86mg),总收率15%,化合物5其结构如下:
[0072][0073]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.30(s,1h),8.99(d,j=4.4hz,1h),8.88(s,1h),8.56(td,j=8.7,3.4hz,2h),8.16-8.11(m,1h),7.89(ddd,j=8.4,6.8,1.5hz,1h),7.83-7.76(m,2h),7.45(d,j=5.4hz,1h),1.68(s,6h).ms:348.87(m
).
[0074]
实施例6
[0075]
2-((3-(2-氯-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物6)合成,将4-溴-2-氯吡啶(384.88mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物6(43mg),总收率8%,化合物6其结构如下:
[0076][0077]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.71(s,1h),8.52(dd,j=5.3,1.9hz,2h),7.75-7.70(m,1h),7.59(dd,j=5.1,1.4hz,1h),7.40(d,j=5.5hz,1h),1.62(s,6h).ms:332.80(m
).
[0078]
实施例7
[0079]
1-((3-(吡啶-4-基乙炔基)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物7)合成,将1-溴环丁烷甲酸乙酯(420.35mg,2.03mmol)替代实施例1中2-溴异丁酸乙酯,其他步骤与实施例1完全相同,得到化合物7(66mg),总收率13%,化合物7其结构如下:
[0080][0081]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.30(s,1h),8.67(d,j=20.1hz,3h),8.45(s,1h),7.58(s,2h),7.09(s,1h),2.90(s,2h),2.44-1.77(m,4h).ms:310.74(m
).
[0082]
实施例8
[0083]
1-((3-(2-甲基吡啶-4-基乙炔基)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物8)合成,将4-溴-2-甲基吡啶(376.04mg,2.00mmol)替代实施例1中4-溴吡啶,将1-溴环丁烷甲酸乙酯(420.35mg,2.03mmol)替代实施例1中2-溴异丁酸乙酯,其他步骤与实施例1完全相同,得到化合物8(56mg),总收率11%,化合物8其结构如下:
[0084][0085]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.25(s,1h),8.63(s,1h),8.54(d,j=5.1hz,1h),8.44(d,j=5.5hz,1h),7.45(s,1h),7.37(dd,j=5.0,1.6hz,1h),7.08(d,j=5.5hz,1h),2.90(ddd,j=12.6,9.1,7.0hz,2h),2.51-2.49(m,3h),2.32(ddd,j=12.7,8.2,4.9hz,2h),2.26-1.97(m,2h).ms:324.86(m
).
[0086]
实施例9
[0087]
1-((3-(3-甲基吡啶-4-基乙炔基)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物9)合成,将4-溴-3-甲基吡啶(376.04mg,2.00mmol)替代实施例1中4-溴吡啶,将1-溴环丁烷甲酸乙酯(420.35mg,2.03mmol)替代实施例1中2-溴异丁酸乙酯,其他步骤与实施例1完全相同,得到化合物9(36mg),总收率7%,化合物9其结构如下:
[0088][0089]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.63(d,j=26.1hz,3h),8.47(d,j=26.5hz,2h),7.52(s,1h),7.14(d,j=5.3hz,1h),2.90(ddd,j=11.7,8.9,6.4hz,2h),2.42(m,3h),2.23(dtt,j=28.8,8.7,3.6hz,3h),2.01(tq,j=10.0,4.9,4.3hz,1h).ms:324.67(m
).
[0090]
实施例10
[0091]
2-((3-(3-氯吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物10)合成,将4-溴-3-氯吡啶(384.88mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物10(58mg),总收率11%,化合物10其结构如下:
[0092][0093]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.81(s,1h),8.70(s,1h),8.61(d,j=4.9hz,1h),8.50(d,j=5.5hz,1h),7.70(d,j=4.9hz,1h),7.44(d,j=5.5hz,1h),1.61(s,6h).ms:332.80(m
).
[0094]
实施例11
[0095]
2-((3-(3-甲氧基吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物11)合成,将4-溴-3-甲氧基吡啶(376.04mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物11(33mg),总收率6%,化合物11其结构如下:
[0096][0097]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.64(s,1h),8.50(d,j=14.8hz,2h),8.28(d,j=4.9hz,1h),7.54(d,j=2.8hz,1h),7.49(d,j=4.7hz,1h),7.36(d,j=5.5hz,1h),4.00(s,3h),1.63(s,6h).ms:328.89(m
).
[0098]
实施例12
[0099]
2-((3-(3,5-二甲基吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物12)合成,将4-溴-3,5-甲基吡啶(372.10mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物12(36mg),总收率14%,化合物12其结构如下:
[0100]
[0101]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.25(s,1h),8.75(s,1h),8.51(d,j=5.4hz,1h),8.40(s,2h),7.39(d,j=5.5hz,1h),2.48(s,6h),1.62(s,6h).ms:326.69(m
).
[0102]
实施例13
[0103]
1-((3-(2-氯吡啶-4-基乙炔基)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物13)合成,将4-溴-3-氯吡啶(384.88mg,2.00mmol)实施例1中4-溴吡啶,将1-溴环丁烷甲酸乙酯(420.35mg,2.03mmol)替代实施例1中2-溴异丁酸乙酯,其他步骤与实施例1完全相同,得到化合物13(63mg),总收率12%,化合物13其结构如下:
[0104][0105]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.26(s,1h),8.66(s,1h),8.49(dd,j=27.0,5.3hz,2h),7.75(d,j=1.3hz,1h),7.61(dd,j=5.1,1.4hz,1h),7.09(d,j=5.5hz,1h),2.97-2.82(m,2h),2.32(ddd,j=12.8,8.3,4.8hz,2h),2.27-1.99(m,2h).ms:344.90(m
).
[0106]
实施例14
[0107]
1-((3-(2-甲氧基吡啶-4-基乙炔基)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物14)合成,将4-溴-2-甲氧基吡啶(376.04mg,2.00mmol)替代实施例1中4-溴吡啶,将1-溴环丁烷甲酸乙酯(420.35mg,2.03mmol)替代实施例1中2-溴异丁酸乙酯,其他步骤与实施例1完全相同,得到化合物14(48mg),总收率9%,化合物14其结构如下:
[0108][0109]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.23(s,1h),8.63(s,1h),8.44(d,j=5.5hz,1h),8.27(d,j=5.2hz,1h),7.16(dd,j=5.2,1.4hz,1h),7.08(d,j=5.5hz,1h),7.01(s,1h),3.89(s,3h),2.99-2.77(m,2h),2.31(ddd,j=10.3,7.8,4.8hz,2h),2.25-1.96(m,2h).ms:340.99(m
).
[0110]
实施例15
[0111]
2-((3-(2-氟吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物15)合成,将4-溴-2-氟吡啶(349.88mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物15(55mg),总收率10%,化合物15其结构如下:
[0112][0113]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.27(s,1h),8.72(s,1h),8.53(d,j=5.5hz,1h),8.35(d,j=5.1hz,1h),7.53(dt,j=5.2,1.6hz,1h),7.43
–
7.38(m,2h),1.63(s,6h).ms:316.07(m
).
[0114]
实施例16
[0115]
2-((3-(3,5-二氯吡啶-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物16)合
成,将4-溴-3,5-二氯吡啶(449.74mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物16(68mg),收率9%,化合物16其结构如下:
[0116][0117]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.77(d,j=30.7hz,3h),8.56(d,j=5.4hz,1h),7.42(d,j=5.5hz,1h),1.62(s,6h).ms:366.00(m
).
[0118]
实施例17
[0119]
2-((3-(1-氟苯基-4-基乙炔基)吡啶-4-基)巯基)-2-甲基丙酸(化合物17)合成,将4-溴氟苯(349.98mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物17(69mg),收率13%,化合物17其结构如下:
[0120][0121]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.29(s,1h),8.64(s,1h),8.45(d,j=5.4hz,1h),7.68
–
7.63(m,2h),7.37
–
7.29(m,3h),1.62(s,6h).ms:315.07(m
).
[0122]
反应通式如下:
[0123][0124]
实施例18
[0125]
中间体a2的合成
[0126][0127]
向50ml的反应瓶中加入3-溴-4氯吡啶(500mg,2.60mmol),硫化钠(304.14mg,3.90mmol),dmf(3ml),反应液在80℃下搅拌4h。反应完毕后,加入20ml水,用1mol
·
l-1
的稀盐酸调节ph至5~6,析出淡黄色固体,抽滤得到中间体a2(385mg),收率77%。
[0128]
中间体b2的合成
[0129][0130]
向50ml的反应瓶中加入中间体a2(500mg,2.63mmol),2-溴异丁酸乙酯(1.03g,5.26mmol),碳酸铯(1.52g,7.89mmol),dmf(5ml),反应液在60℃下搅拌4h。反应完毕后,加入20ml水,用乙酸乙酯萃取三次(3
×
50ml),有机层合并,饱和食盐水洗,无水硫酸钠干燥,过滤,旋蒸后过柱得中间体b2(565mg),收率70%。
[0131]
中间体d2的合成
[0132][0133]
向50ml反应瓶中加入中间体b2(400mg,1.31mmol),2-甲基-3-丁炔-2-醇c2(276.52mg,3.29mmol),三苯基膦(68.98mg,0.26mmol),碘化亚铜(25.04mg,0.13mmol),碳酸钾545.18mg,3.94mmol),pd/c(400mg)和dmf(5ml),氮气置换3次后,氮气保护下80℃反应8h。反应完毕后,反应液硅藻土助滤,甲醇洗涤滤饼,滤液旋蒸后过柱得中间体d2(255mg),收率62%。
[0134]
2-((3-2-甲基-3-丁炔-2-醇)吡啶-4-基)巯基)-2-甲基丙酸e2(化合物18)合成
[0135][0136]
向50ml的反应瓶中加入中间体d2(150mg,0.49mmol),用2mol
·
l-1
lioh水溶液(2.50ml,5.00mmol),甲醇(10ml),室温反应5h。反应完毕后,反应液旋干,加入10ml水溶解后过滤,滤液用1n盐酸中和至ph为5-6,析出固体,过滤,滤饼烘干得化合物18(90mg),收率66%。1h nmr(400mhz,dmso-d6)δ8.28(s,1h),8.20(d,j=5.5hz,1h),7.59(d,j=5.5hz,1h),1.49(d,j=4.9hz,12h).ms:279.53(m
).
[0137]
实施例19
[0138]
2-((3-戊-1丁炔-3-醇)吡啶-4-基)巯基)-2-甲基丙酸(化合物19)合成,将戊-1丁炔-3-醇(276.52mg,3.29mmol)替代实施例18中2-甲基-3-丁炔-2-醇,其他步骤与实施例18完全相同,得到化合物19(95mg),收率24%,化合物19其结构如下:
[0139][0140]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.45(s,1h),8.39(d,j=5.4hz,1h),7.25(d,j=5.5hz,1h),1.60(s,6h),1.49(s,5h).ms:279.79(m
).
[0141]
实施例20
[0142]
2-((3-丁基-3-丁炔-2-醇)吡啶-4-基)巯基)-2-甲基丙酸(化合物20)合成,将2-甲基-3-丁炔-2-醇用丁基-3-丁炔-2-醇(230.59mg,3.29mmol)替代实施例18中2-甲基-3-丁炔-2-醇,其他步骤与实施例18完全相同,得到化合物20(83mg),收率19%,化合物20其结构如下:
[0143][0144]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.47(s,1h),8.40(d,j=5.4hz,1h),7.25(d,j=5.5hz,1h),1.61(s,6h),1.41(d,j=6.6hz,3h).ms:265.93(m
).
[0145]
实施例21
[0146]
1-((3-2-甲基-3-丁炔-2-醇)吡啶-4-基)巯基)-1-环丁烷丙酸(化合物21)合成,
将1-溴环丁烷甲酸乙酯(1.08g,5.26mmol)替代实施例18中2-甲基-3-丁炔-2-醇,其他步骤与实施例18完全相同,得到化合物21(92mg),收率23%,化合物21其结构如下:
[0147][0148]
表征结果如下:1h nmr(300mhz,dmso-d6)δ8.42(s,1h),8.36(d,j=5.5hz,1h),7.03(d,j=5.5hz,1h),5.90-5.42(m,1h),2.91(dd,j=10.7,4.6hz,2h),2.26(d,j=6.0hz,2h),1.54(s,8h).ms:291.83(m
).
[0149]
实施例22
[0150]
2-甲基-2-(((3-(苯基乙炔基)吡啶-4-基)硫基)丙酸(化合物22)合成,将苯基乙炔基三甲基硅烷(348.63mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物22(62mg),总收率20%,化合物22结构如下:
[0151][0152]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.25(s,1h),8.65(s,1h),8.46(d,j=5.4hz,1h),7.60(dtdd,j=7.4,5.7,4.4,1.8hz,2h),7.52
–
7.46(m,3h),7.34(d,j=5.4hz,1h),1.63(s,6h).ms:297.82(m
).
[0153]
实施例23
[0154]
2-甲基-2-((3-(对甲苯基乙炔基)吡啶-4-基)硫基)丙酸(化合物23)合成,将4-溴甲苯(342.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物23(90mg),总收率25%,化合物23结构如下:
[0155][0156]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.50(s,1h),8.30(d,j=5.5hz,1h),7.55(d,j=5.5hz,1h),7.46(d,j=8.0hz,2h),7.27(d,j=7.9hz,2h),2.35(s,3h),1.53(d,j=11.7hz,6h).ms:311.90(m
).
[0157]
实施例24
[0158]
2-甲基-2-((3-(间甲苯基乙炔基)吡啶-4-基)硫基)丙酸(化合物24)合成,将4-溴3-甲基苯(342.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物24(78mg),总收率22%,化合物24结构如下:
[0159][0160]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.58(s,1h),8.39(d,j=5.5hz,1h),7.45
–
7.33(m,4h),7.28(d,j=7.3hz,1h),2.34(s,3h),1.60(s,6h).ms:311.90(m
).
[0161]
实施例25
[0162]
2-甲基-2-((3-(邻甲苯基乙炔基)吡啶-4-基)硫基)丙酸(化合物25)合成,将4-溴2-甲基苯(342.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物25(86mg),总收率24%,化合物25结构如下:
[0163][0164]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.27(s,1h),8.62(s,1h),8.43(d,j=5.4hz,1h),7.55
–
7.48(m,2h),7.36
–
7.28(m,3h),1.62(s,6h),1.20(t,j=7.6hz,3h).ms:311.90(m
).
[0165]
实施例26
[0166]
2-((3-((4-乙基苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物26)合成,将4-溴乙基苯(342.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物26(71mg),总收率21%,化合物26结构如下:
[0167][0168]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.65(s,1h),8.44(d,j=5.4hz,1h),7.57
–
7.53(m,1h),7.39
–
7.34(m,3h),7.28(dt,j=7.7,2.6hz,1h),2.52(s,3h),2.51
–
2.50(m,2h),1.62(s,6h).ms:325.90(m
).
[0169]
实施例27
[0170]
2-((3-((2,4-二甲基苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物27)合成,将4-溴-2,4-二甲基苯(342.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物27(62mg),总收率20%,化合物27结构如下:
[0171][0172]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.45(s,1h),8.33
–
8.19(m,1h),7.78
–
7.64(m,1h),7.40(d,j=7.7hz,1h),7.23
–
7.01(m,2h),2.48(s,3h),2.31(d,j=8.6hz,3h),1.50(s,6h).ms:325.90(m
).
[0173]
实施例28
[0174]
2-((3-((3,5-二甲基苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物28)合成,将4-溴-3,5-二甲基苯(342.07mg,
[0175]
2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物28(82mg),总收率26%,化合物28结构如下:
[0176]
[0177]
表征结果如下:1h nmr(500mhz,dmso-d6)δ8.45(s,1h),8.25(d,j=5.4hz,1h),7.67(d,j=5.3hz,1h),7.18(s,2h),7.09(s,1h),2.30(s,6h),1.53(s,6h).ms:325.90(m
).
[0178]
实施例29
[0179]
2-((3-((3-氟苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物29)合成,将间溴氟苯(350.00mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物29(78mg),总收率24%,化合物29结构如下:
[0180][0181]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.11(s,1h),8.53(s,1h),8.34(d,j=5.5hz,1h),7.39(td,j=7.7,5.7hz,1h),7.34
–
7.28(m,2h),7.26
–
7.18(m,2h),1.49(s,6h).ms:315.86(m
).
[0182]
实施例30
[0183]
2-((3-((2-氟苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物30)合成,将邻溴氟苯(350.00mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物30(72mg),总收率22%,化合物30结构如下:
[0184][0185]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.31(s,1h),8.66(s,1h),8.56
–
8.39(m,1h),7.67(td,j=7.5,1.8hz,1h),7.54(tdd,j=7.7,5.4,1.8hz,1h),7.38(dd,j=11.5,7.0hz,2h),7.32(t,j=7.6hz,1h),1.62(s,6h).ms:315.86(m
).
[0186]
实施例31
[0187]
2-((3-((4-甲氧基苯基)乙炔基)吡啶-4-基)硫基)-2-甲基丙酸(化合物31)合成,将4-溴苯甲醚(374.07mg,2.00mmol)替代实施例1中4-溴吡啶,其他步骤与实施例1完全相同,得到化合物31(62mg),总收率20%,化合物31结构如下:
[0188][0189]
表征结果如下:1h nmr(500mhz,dmso-d6)δ13.22(s,1h),8.60(s,1h),8.42(d,j=5.5hz,1h),7.57
–
7.50(m,2h),7.32(d,j=5.4hz,1h),7.09
–
7.00(m,2h),3.81(s,3h),1.62(s,6h).ms:327.90(m
).
[0190]
实施例32
[0191]
化合物1~31的体外活性测试
[0192]
体外的urat1试验可用于鉴别对降低血清尿酸具有潜在活性的化合物。适宜的试验包括对细胞(人类胚胎肾细胞,hek293:中国科学院细胞库gnhu18)进行转染稳定表达的慢病毒载体构建,获得转染细胞-hek293/hurat1细胞,经转染的hek293/hurat1细胞将被用
于14c-尿酸转运活性的测试。以作为urat1抑制剂的化合物阻断转染细胞摄取尿酸的能力来评估所述化合物的活性。本发明化合物的hurat1生化抑制活性通过以下的试验进行测定,测得的ic
50
值。
[0193]
将hek293/hurat1细胞在emem培养基中以1
×
105细胞/孔的密度接种于涂有多聚d-赖氨酸的96孔板(becton dickinson,货号356509)中,并孵育过夜。在汉克斯平衡盐溶液(hbss)中使用或不使用测试化合物来制备含有最终浓度为11.57μm的14c-尿酸(american radioactive compound,货号arc 0513a)的反应溶液,所述汉克斯平衡盐溶液(hbss)包含125mm葡萄糖酸钠、4.8mm葡萄糖酸钾、1.2mm磷酸二氢钾、1.2mm硫酸镁、1.3mm葡萄糖酸钙、5.6mm葡萄糖和25mm hepes(ph7.3)。使用洗涤缓冲液(125mm葡萄糖酸钠、10mm hepes,ph7.3)进行一次洗涤来冲洗培养基后,将制备的反应溶液添加至各孔中,并在室温孵育12分钟。然后移除反应溶液,以洗涤缓冲液洗涤细胞两次,并用0.2m naoh裂解5分钟。将细胞裂解液转移到含闪烁液的96孔培养板(perkinelmer,货号1450-401)中,并在microbeta计数器(perkinelmer)中对放射性进行计数。
[0194]
将测试化合物溶解在dmso中,然后将相同浓度的dmso加入不包含测试化合物的hek293/hurat1细胞孔中。将各测试条件下的细胞的尿酸摄取表示为相对dmso对照的平均百分比抑制率。将对包含dmso的孔得到的放射性值视为细胞的100%摄取。化合物的ic
50
值可通过不同浓度下的抑制率计算得出。
[0195]
表1本发明所述化合物对尿酸转运蛋白1(urat1)的活性抑制的ic
50
[0196]
[0197][0198]
结果显示,受试化合物与阳性药物lesinurad和苯溴马隆相比较,其中:a表示ic
50
值在1nm至200nm的范围;b表示ic
50
值在200nm至1μm的范围;c表示ic
50
值大于1μm。
[0199]
根据如上表中列出的实验数据可以看出,本发明的化合物相对于已在临床应用药物lesinurad和苯溴马隆具有更佳或相似的ic
50
值,由此表明本发明的所述化合物具有较好的抑制尿酸重吸收的活性,可以作为新型高效降低血液尿酸的药物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。