一种采用hplc-pda检测板青败毒口服液中13种有效成分的方法
技术领域
1.本发明涉及一种采用hplc-pda检测板青败毒口服液中13种有效成分的方法,属于兽药中有效成分的检查方法。
背景技术:
2.板青败毒口服液现收载于《兽药质量标准》2017年版中药卷,由金银花、大青叶、板蓝根、蒲公英、白英、连翘、甘草、天花粉、白芷、防风、赤芍、浙贝母共12味药提取精制而成的口服溶液剂,现标准对金银花、蒲公英(绿原酸)、甘草(甘草酸铵)、板蓝根(精氨酸)、赤芍(芍药苷)分别进行了薄层鉴别。本方法存在很大的缺点:薄层鉴别展开剂共采用乙酸丁酯、甲酸、正丁醇、冰醋酸、乙酸乙酯、三氯甲烷等多种有机溶剂,污染大、前处理繁琐,展开耗时长,结果不易判定。
技术实现要素:
3.本发明针对上述问题,提供了一种采用hplc-pda检测板青败毒口服液中13种有效成分的方法,本发明采用反相高效液相色谱(uplc-pda)分离技术,对板青败毒液口服中有效成分金银花、蒲公英、板蓝根、大青叶、甘草、赤芍、连翘等开展一法多测研究,优化提取方法、色谱条件,尤其是需要优化检测波长和梯度洗脱程序,使尽可能多的目标峰分离出来,对含量较高的组分进行含量测定,实现定性或定量,更高效、快速、环保。
4.本发明成功建立了一种采用hplc-pda检测板青败毒口服液中13种有效成分的方法,其中,(r,s)-告依春来检测板蓝根,大青叶;新绿原酸、单咖啡酰酒石酸、隐绿原酸、绿原酸、咖啡酸、菊苣酸和4,5-二-o-咖啡酰奎宁酸来检测金银花和蒲公英;连翘酯苷、连翘苷来检测连翘;芍药苷来检测赤芍;甘草酸和甘草苷来检测甘草。
5.进一步的,本发明同时鉴别板青败毒口服液中多种成分的方法,包括下述步骤:
6.第一步:供试品溶液的制备:取供试品至棕色容量瓶,加入溶剂,定容至25ml,振摇后超声提取20-40min,过滤,作为供试品溶液;所述溶剂选自10%甲醇、50%甲醇、20%乙醇、50%乙醇或者75%乙醇(体积分数);优选为20%的乙醇。
7.优选的,供试品的取样量为1ml;提取时间30min;
8.第二步,对照品溶液的制备:各取(r,s)-告依春、新绿原酸、单咖啡酰酒石酸、隐绿原酸、绿原酸、芍药苷、咖啡酸、甘草苷、连翘酯苷、连翘苷、菊苣酸、4,5-二-o-咖啡酰奎宁酸和甘草酸对照品,分别加无水乙醇或甲醇或75%乙醇,制备对照品储备溶液;再取13种储备液混合,作为混合对照品储备溶液;
9.第三步:液相色谱法:hplc-pda检测器;3d扫描波长:190-400nm,2d检测波长240nm,230nm,251nm,327nm,330nm;2d检测波长优选为240nm。
10.色谱柱:英国ace c18-amide色谱柱,150*4.6mm粒径3μm;
11.进样量1-5μl;
12.柱温30-40度,优选为35度;
13.流动相a项:甲醇;流动相b项:0.4%磷酸溶液;
14.流速:0.72ml/min;
15.梯度洗脱程序:
16.0-16min,a相保持20%,b相保持80%;
17.16-17min,a相由20%升至30%,b相由80%降至70%;
18.17-25min,a相保持30%,b相保持70%
19.25-28min,a相由30%升至33%,b相由70%降至67%;
20.28-33min,a相保持33%,b相保持67%;
21.33-43min,a相由33%升至45%,b相由67%降至55%;
22.43-49min,a相保持45%,b相保持55%;
23.49-52min,a相由45%升至75%,b相由55%降至25%;
24.52-56min,a相保持75%,b相保持25%;
25.56-58min,a相由75%降至20%,b相由25%升至80%;
26.58-60min,a相保持20%,b相保持80%。
27.本发明与现有技术相比具有以下优点:
28.本发明通过一套液相色谱系统同时把13种目标化合物一次鉴别出来,方法简便快速、分离效果好,鉴别精度高,结果重现性好,易于观察,专属性强,色谱峰峰形良好,既达到中国兽药典的要求,还节省时间、试剂。
附图说明
29.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定
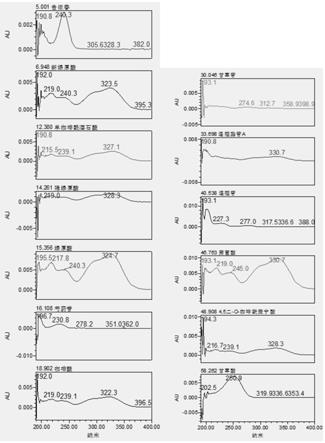
30.图1为13种混合对照品光谱图;
31.图2为13种混合对照品色谱图,240nm;
32.图3为实施例1中采用本发明所述方法10%甲醇提取做出的板青败毒口服液的液相色谱图,240nm
33.图4为实施例1中采用本发明所述方法50%甲醇提取做出的板青败毒口服液的液相色谱图,240nm;
34.图5为实施例1中采用本发明所述方法20%乙醇提取做出的板青败毒口服液的液相色谱图,240nm;
35.图6为实施例1中采用本发明所述方法50%乙醇提取做出的板青败毒口服液的液相色谱图,240nm;
36.图7为实施例1中采用本发明所述方法75%乙醇提取做出的板青败毒口服液的液相色谱图,240nm。
37.图8为13种混合对照品色谱图,250nm;
38.图9为13种混合对照品色谱图,327nm;
39.图10为13种混合对照品色谱图,330nm;
40.图11为13种混合对照品色谱图,230nm;
41.图12为实施例1中采用本发明所述方法20%乙醇提取做出的板青败毒口服液的液相色谱图,327nm。
42.图13为实施例1中采用本发明所述方法20%乙醇提取做出的板青败毒口服液的液相色谱图,251nm。
43.图14为实施例1中采用本发明所述方法20%乙醇提取做出的板青败毒口服液的液相色谱图,230nm。
44.图15为实施例1中采用本发明所述方法20%乙醇提取做出的板青败毒口服液的液相色谱图,330nm。
具体实施方式
45.下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
46.实施例1:一种采用hplc-pda检测板青败毒口服液中13种有效成分的方法
47.其中,(r,s)-告依春来检测板蓝根和大青叶;新绿原酸、单咖啡酰酒石酸、隐绿原酸、绿原酸、咖啡酸、菊苣酸和4,5-二-o-咖啡酰奎宁酸来检测金银花和蒲公英;连翘酯苷、连翘苷来检测连翘;芍药苷来检测赤芍;甘草酸和甘草苷来检测甘草;
48.包括下述步骤:
49.第一步:供试品溶液的制备:取供试品1ml至棕色容量瓶,加不同浓度的乙醇/甲醇溶液(见表1),定容至25ml,振摇后超声提取,提取时间20-40min,过滤,作为供试品溶液;
50.表1不同的提取溶剂表
51.序号溶剂提取时间min色谱图见110%甲醇40图3250%甲醇20图4320%乙醇30图5、图8-图15450%乙醇40图6575%乙醇20图7
52.第二步,对照品溶液的制备:另各取(r,s)-告依春、新绿原酸、单咖啡酰酒石酸、隐绿原酸、绿原酸、芍药苷、咖啡酸、甘草苷、连翘酯苷、连翘苷、菊苣酸、4,5-o-二咖啡酰奎宁酸和甘草酸对照品适量至容量瓶,分别加无水乙醇或甲醇或75%乙醇,制备对照品储备溶液;再取13种储备液适量体积混合,作为混合对照品储备溶液:对照品储备溶溶液和混合对照品溶液的配制见表2-1、表2-2和表2-3,其混合对照品的光谱图和色谱图见图1-图2,根据保留时间和光谱图的峰形不同,来判断待测样品中的目标化合物。
53.表2-1前7种对照品混合体积比
[0054][0055]
表2-2 2种对照品混合体积比
[0056][0057]
表2-3 2种对照品混合体积比
[0058][0059]
前7种混合对照取8ml 甘草酸甘草苷混合溶液2ml 连翘苷连翘酯苷混合溶液2ml 芍药苷1.5ml 表告依春2ml 20%乙醇0.5ml得13种混合对照品溶液。最终浓度如表2-4。
[0060]
表2-4
[0061][0062]
第三步:液相色谱法:hplc-pda检测器;3d扫描波长:190-400nm,2d检测波长240nm,324nm,327nm,230nm,250nm,330nm;色谱柱:英国ace c18-amide色谱柱,150*4.6mm粒径3μm。进样量1-5μl。优选柱温35度;流动相a项:甲醇;流动相流动相:流动相b项:0.4%磷酸溶液;流速0.72ml/min;按表3中的规定进行梯度洗脱。结果判定:根据色谱图,供试品色谱中,在与13种对照品色谱相应的位置上,出现相同的色谱峰,且光谱图一致,表明可检出相应目标化合物;并通过对照品和样品色谱峰面积的比值来计算并进行定量。
[0063]
表3梯度洗脱条件表
[0064][0065]
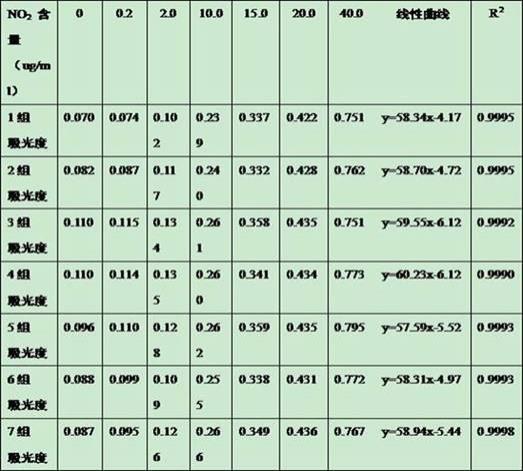
所述第三步的检测波长是217nm,240nm,324nm,327nm,230nm,250nm,330nm等。由于观察217nm色谱图干扰较大,我们最初又选择327nm,新绿原酸、单咖啡酰酒石酸、隐绿原酸、绿原酸、咖啡酸、菊苣酸、4,5-二-o-咖啡酰奎宁酸等响应值非常高,但芍药苷、连翘苷、甘草酸和表告依春相应非常低,几乎检测不到,而且由于表告依春的含量比较低,我们选择表告依春的最大吸收波长240nm,作为最佳波长。而且13种化合物响应值均较高,背景干扰小。色谱结果如表4-表16。
[0066]
表4 4,5-二-o-咖啡酰奎宁酸色谱结果表
[0067][0068]
表5单咖啡酰酒石酸结果表
[0069][0070]
表6甘草酸结果表
[0071][0072]
表7甘草苷色谱结果表
[0073][0074]
表8(r,s)-告依春色谱结果表(由于含量太少,含量单位为μg/ml)
[0075][0076]
表9菊苣酸色谱结果表
[0077][0078]
表10咖啡酸色谱结果表
[0079][0080]
表11连翘苷色谱结果表
[0081][0082]
表12连翘酯苷a色谱结果表
[0083][0084]
表13绿原酸色谱结果表
[0085][0086]
表14芍药苷色谱结果表
[0087][0088]
表15新绿原酸色谱结果图
[0089][0090]
表16隐绿原酸色谱结果图
[0091][0092]
3d扫描范围190-400nm,2d检测波长选择217nm,240nm,324nm,327nm,230nm,250nm,330nm等。由于(r,s)-告依春最大吸收波长240nm,在251nm、324nm、327nm、330nm处几乎无吸收,最终采用240nm来采集。
[0093]
各种化合物usp拖尾因子在0.95-1.15范围内,理论塔板数在10000-1000000范围内,响应良好。本发明同时针对板青败毒口服液中对13种目标化合物进行一法多测研究,完全满足检测需求,提升检测效率,降低检测污染,保护检验人员。
[0094]
本发明方法对现有的质量标准进行提升,为制定2025版中华人民共和国兽药典标准,提供检测基础和条件。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。